GROMACS 安装:详细教程来袭
前言
GROMACS 是一款功能强大的开源分子动力学模拟软件,广泛应用于生物化学、材料科学和物理学等领域。它支持多种模拟类型,包括能量最小化、分子动力学模拟和自由能计算等。本文将为大家介绍在使用西柚云GPU服务器的过程中,如何根据显卡的不同型号(30系、40系和50系)安装 GROMACS。
GROMACS安装
CPU版本
sudo apt update
sudo apt install gromacs
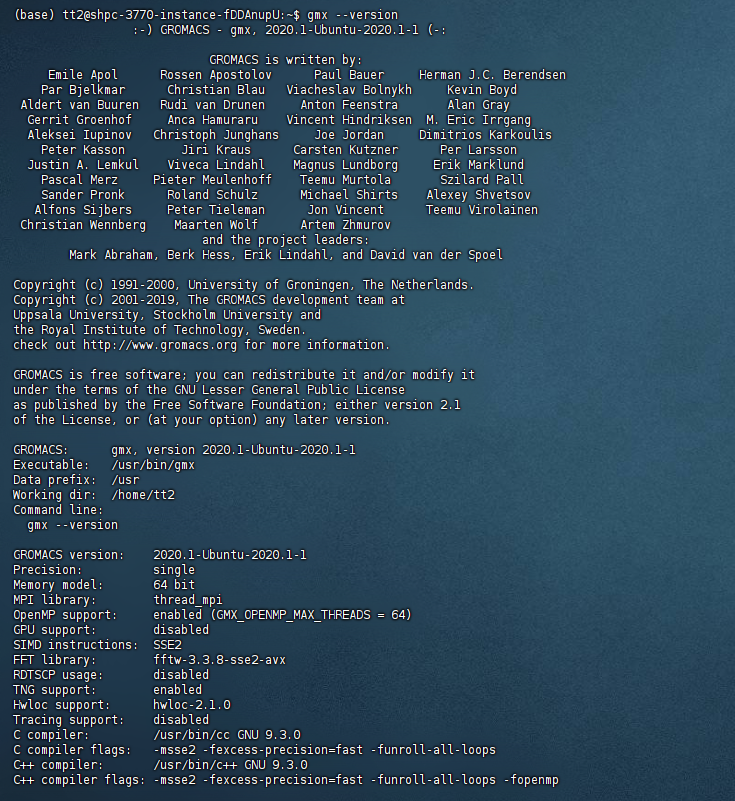
GPU版本
- Gromacs(GPU版)需要从源代码编译安装,先根据自己的服务器套餐安装对应的显卡驱动:西柚云GPU服务器使用指南
30系显卡安装步骤
1、检查 CMake 版本
- cmake需要3.21以上。执行cmake --version命令查看cmake版本,如果你的cmake版本低于3.21,可以按照下列步骤更新
# 下载安装脚本
wget http://mirror.xiyoucloud.pro:63332/static/software/cmake-3.21.0-linux-x86_64.sh# 执行安装脚本
sudo bash cmake-3.21.0-linux-x86_64.sh --skip-licence --prefix=/usr# 安装过程中需要确认信息,依次执行
q
y
n
2、安装CUDA12.8
- 下载并安装 CUDA Toolkit
wget https://developer.download.nvidia.com/compute/cuda/repos/ubuntu2004/x86_64/cuda-keyring_1.1-1_all.deb
sudo dpkg -i cuda-keyring_1.1-1_all.deb
sudo apt-get update
sudo apt-get -y install cuda-toolkit-12-8# 将 CUDA 路径添加到环境变量
echo 'export PATH=/usr/local/cuda-12.8/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
3、安装MPI
- 如果不安装的话后续编译gromacs会出现下图报错
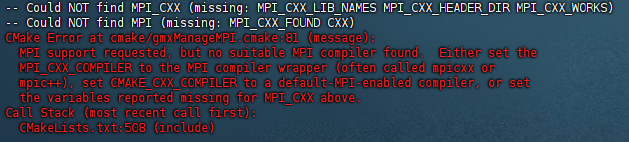
sudo apt update
sudo apt install openmpi-bin libopenmpi-dev -y# 验证 MPI 是否安装成功
mpicxx --version
4、安装 GROMACS-2023.2
- 30系显卡需要安装gromacs2023-2版本
# 下载gromacs-2023.2,解压后进入解压目录
wget ftp://ftp.gromacs.org/pub/gromacs/gromacs-2023.2.tar.gz
tar -xzf gromacs-2023.2.tar.gz
cd gromacs-2023.2# 创建构建目录并编译安装
mkdir build && cd build
cmake .. \-DGMX_BUILD_OWN_FFTW=ON \-DGMX_GPU=CUDA \-DGMX_CUDA_TARGET_SM=86 \-DGMX_MPI=ON \-DGMX_OPENMP=ON \-DCMAKE_INSTALL_PREFIX=/home/用户名/gromacs_install
make -j$(nproc)
make install# 设置环境变量并创建符号链接,记得修改为自己的用户名
echo 'source /home/用户名/gromacs_install/bin/GMXRC' >> ~/.bashrc
source ~/.bashrc
ln -s /home/用户名/gromacs_install/bin/gmx_mpi /home/用户名/gromacs_install/bin/gmx
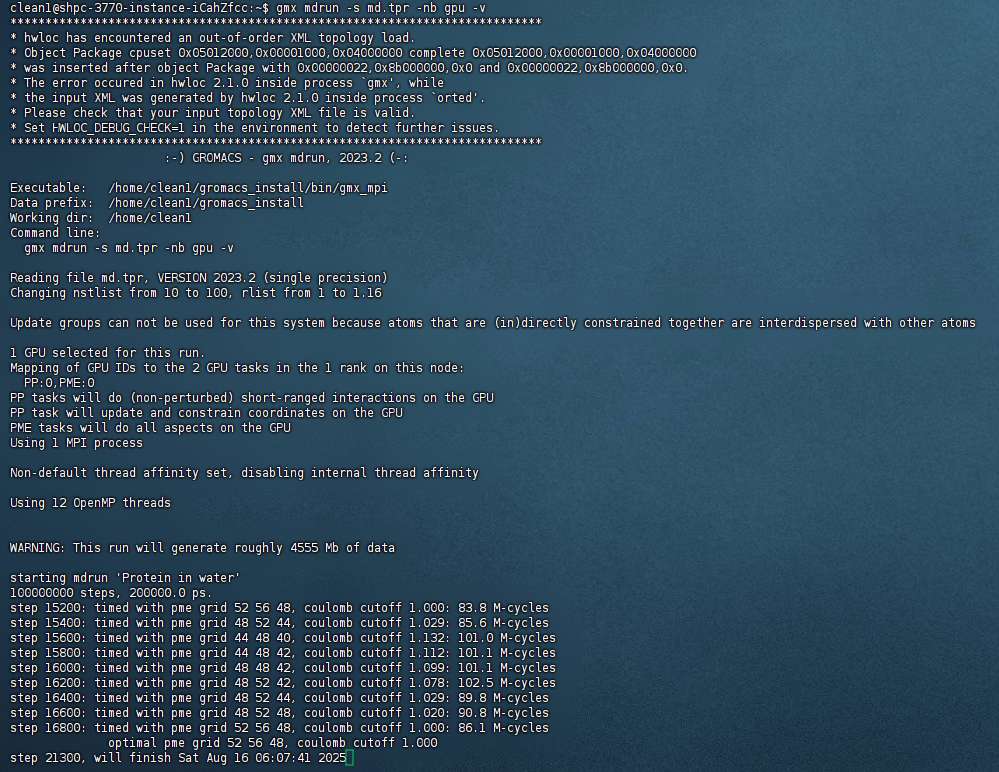
- 命令测试
gmx mdrun -s md.tpr -nb gpu -v
40系、50系显卡安装步骤
1、检查 CMake 版本
- 40系、50系显卡可以安装gromacs2025-2版本,其cmake需要3.28版本以上。执行cmake --version命令查看cmake版本,如果你的cmake版本低于3.28,可以按照下列步骤更新
# 下载安装脚本
wget http://mirror.xiyoucloud.pro:63332/static/software/cmake-3.28.0-linux-x86_64.sh# 执行安装脚本
sudo bash cmake-3.28.0-linux-x86_64.sh --skip-licence --prefix=/usr# 安装过程中需要确认信息,依次执行
q
y
n
2、安装gcc 11和g++11
- gromacs2025-2版本的编译需要gcc/g++11,执行gcc --version和g++ --version命令查看版本,如果不是gcc/g++11,可以按照下列步骤更新
sudo apt update
sudo apt install software-properties-common
sudo add-apt-repository ppa:ubuntu-toolchain-r/test
sudo apt update
sudo apt install gcc-11 g++-11 -y# 设置gcc11和g++11为默认版本
sudo update-alternatives --install /usr/bin/gcc gcc /usr/bin/gcc-11 60 --slave /usr/bin/g++ g++ /usr/bin/g++-11
3、安装CUDA 12.8
- 下载并安装 CUDA Toolkit
wget https://developer.download.nvidia.com/compute/cuda/repos/ubuntu2004/x86_64/cuda-keyring_1.1-1_all.deb
sudo dpkg -i cuda-keyring_1.1-1_all.deb
sudo apt-get update
sudo apt-get -y install cuda-toolkit-12-8# 将 CUDA 路径添加到环境变量
echo 'export PATH=/usr/local/cuda-12.8/bin:$PATH' >> ~/.bashrc
source ~/.bashrc
4、安装MPI
- 如果不安装的话后续编译gromacs会出现下图报错
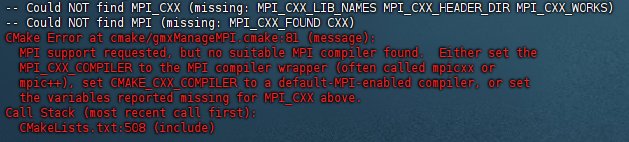
sudo apt update
sudo apt install openmpi-bin libopenmpi-dev -y# 验证 MPI 是否安装成功
mpicxx --version
5、安装 GROMACS-2025.2
# 下载gromacs-2025.2,解压后进入解压目录
wget https://ftp.gromacs.org/pub/gromacs/gromacs-2025.2.tar.gz
tar -xzf gromacs-2025.2.tar.gz
cd gromacs-2025.2
# 创建构建目录并编译安装
mkdir build && cd buildcmake .. \-DGMX_BUILD_OWN_FFTW=ON \-DGMX_GPU=CUDA \-DGMX_CUDA_TARGET_SM=89 \-DGMX_MPI=ON \-DCMAKE_INSTALL_PREFIX=$HOME/gromacs_install \-DCMAKE_C_COMPILER=/usr/bin/gcc \-DCMAKE_CXX_COMPILER=/usr/bin/g++
make -j$(nproc)
make install# 设置环境变量并创建符号链接,记得修改为自己的用户名
echo 'source /home/用户名/gromacs_install/bin/GMXRC' >> ~/.bashrc
source ~/.bashrc
ln -s /home/用户名/gromacs_install/bin/gmx_mpi /home/用户名/gromacs_install/bin/gmx
- 命令测试
gmx mdrun -s md.tpr -nb gpu -v
总结
通过本文,我们希望能够帮助大家更便捷地安装 GROMACS,无论是使用 CPU 还是 GPU ,都能更加得心应手地进行分子动力学模拟。如果大家对我们的内容有任何反馈,或者有特定的软件使用教程需求,欢迎在后台留言告诉我们,我们系统整理后与大家分享。