AbMole| MGCD0103(M1790,Mocetinostat)
Mocetinostat (MGCD0103)是一种有效的HDAC抑制剂,对HDAC1抑制作用最强,IC50为0.15 μM,比作用于HDAC2, 3,和11选择性高2到10倍,对HDAC4, 5, 6, 7,和8没有抑制活性。
一、化学性质/溶解性/储存
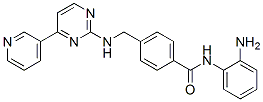
| 分子量 | 396.44 |
| 分子式 | C23H20N6O |
| CAS号 | 726169-73-9 |
| 溶解性 | DMSO 20 mg/mL |
| 储存条件 | 粉末型式 -20°C 3年;4°C 2年 溶于溶剂 -80°C 6个月;-20°C 1个月 |
| 运输方式 | 冰袋运输,根据产品的不同,可能会有相应调整。 |
二、作用机制:选择性HDAC抑制
MGCD0103的核心机制在于选择性抑制I类HDAC(HDAC1、HDAC2、HDAC3)和IV类HDAC(HDAC11),而对II类酶(如HDAC4-8)无明显作用。其抑制活性表现为:
IC50值:对HDAC1的抑制活性最强(0.15 μM),其次是HDAC2(0.29 μM)和HDAC11(0.59 μM),对HDAC3的抑制较弱(1.66 μM)。
结构特异性:其分子中的外环氨基结构是结合HDAC活性位点的关键,通过阻断乙酰化组蛋白的脱乙酰过程,诱导染色质重构,从而调控基因表达。
这种选择性抑制不仅减少了对非靶标HDAC的脱靶效应,还降低了传统泛HDAC抑制剂(如伏立诺他)常见的血液学毒性风险。
三、生物活性
Mocetinostat (MGCD0103)是一种有效的HDAC抑制剂,对HDAC1抑制作用最强,IC50为0.15 μM,比作用于HDAC2, 3,和11选择性高2到10倍,对HDAC4, 5, 6, 7,和8没有抑制活性。MGCD0103按剂量依赖性在纳摩尔浓度或低微摩尔浓度时只能抑制9个人类重组HDACs中的一部分,包括HDAC1, HDAC2, HDAC3, 和HDAC11。在体外,MGCD0103有效抑制人类HDAC1和HDAC2酶,但是不抑制二级HDACs。MGCD0103的外环氨基抑制酶时是必需的,因为作用于HDAC1和HDAC2的HDAC抑制活性已被desamino类似物全部消除。
四、临床研究进展
1. 适应症与孤儿药资格
MGCD0103目前主要针对以下疾病开展研究:
血液肿瘤:弥漫性大B细胞淋巴瘤(DLBCL)、骨髓增生异常综合征(MDS)。
实体瘤:膀胱癌及携带CREBBP/EP300基因突变的肿瘤。美国FDA已授予其治疗DLBCL和MDS的孤儿药资格,以加速其临床开发。
2. 联合治疗策略
化疗协同:与吉西他滨联用可增强对胰腺癌细胞的杀伤作用,机制可能与抑制DNA修复通路相关。
免疫治疗协同:联合PD-L1抑制剂(如Durvalumab)在非小细胞肺癌中进入II期试验,旨在通过增强T细胞活性提升疗效。
3. 临床试验阶段
截至最新研究数据(2025年),MGCD0103尚处于II期临床试验阶段,暂未获批上市。早期试验显示,其在复发/难治性淋巴瘤中的客观缓解率(ORR)为20%-30%,但部分患者出现疲劳、恶心等轻度不良反应。