哈佛团队在Cancer Cell发表多模态医学AI模型,整合病理切片和基因组特征,为癌症预后提供新思路
小罗碎碎念
这篇文章对于我来说,意义独特,因为它是我读研以后,第一次组会汇报时选择的文献,当时的自己读的很懵,现在再拿出来重温一下。
这篇发表于Cancer Cell的论文,利用多模态深度学习整合病理全切片图像和分子特征,为癌症预后预测提供了新的思路和方法。

研究提出一种基于深度学习的多模态融合(MMF)算法,将H&E全切片图像和分子特征相结合,以评估癌症死亡风险。
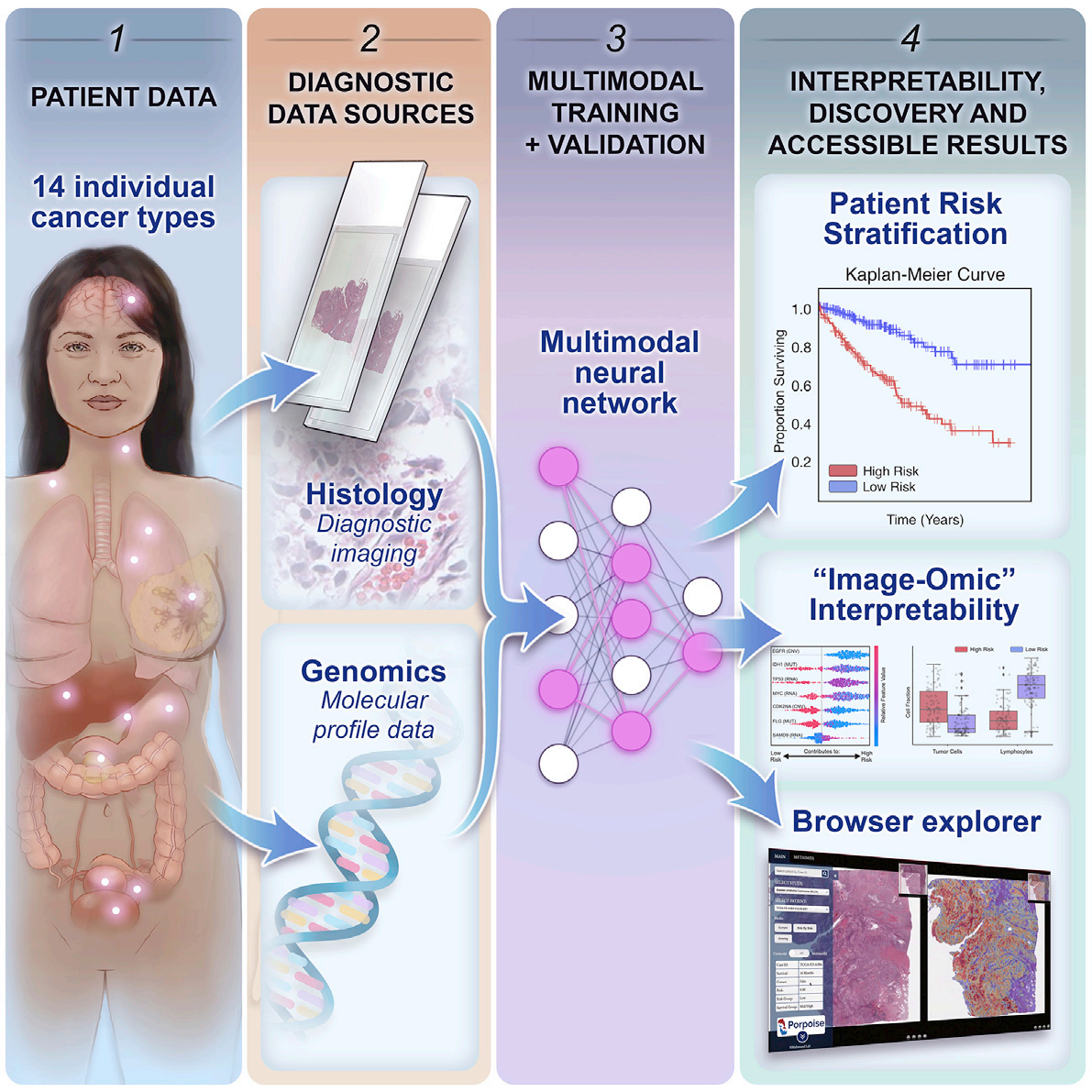
研究对14种癌症类型的5720例患者样本进行分析,结果显示,MMF模型在多数癌症类型中表现更优,能更好地进行患者风险分层。同时,研究还开发了PORPOISE平台,用于可视化和解释模型结果,发现免疫细胞浸润等形态和分子特征与患者预后相关。
该研究为医学AI在癌症预后预测中的应用提供了重要参考,多模态融合的方法展现出良好的应用前景,有望推动癌症精准医疗的发展。但研究也存在一定局限性,未来可进一步优化模型,探索更多潜在的生物标志物,为临床决策提供更有力的支持。
交流群
欢迎大家加入【医学AI】交流群,本群设立的初衷是提供交流平台,方便大家后续课题合作。
目前小罗全平台关注量61,000+,交流群总成员1400+,大部分来自国内外顶尖院校/医院,期待您的加入!!
由于近期入群推销人员较多,已开启入群验证,扫码添加我的联系方式,备注姓名-单位-科室/专业,即可邀您入群。
知识星球
对推文中的内容感兴趣,想深入探讨?在处理项目时遇到了问题,无人商量?加入小罗的知识星球,寻找科研道路上的伙伴吧!
一、文献概述
文章利用多模态深度学习整合 14 种癌症类型的全切片病理图像与分子特征数据,构建预后模型,通过对比单模态模型及分析模型可解释性,发现相关预后特征,开发出 PORPOISE 平台辅助研究,为癌症预后评估提供新方法与思路。
1-1:研究背景
癌症具有组织病理学、基因组和转录组异质性,当前临床评估依赖主观的组织病理特征判断,存在观察者间和观察者内的高变异性,且缺乏对肿瘤微环境中细胞空间组织与患者风险关系的深入理解。
深度学习在计算病理学的应用虽有进展,但现有预后模型多基于单一数据来源,难以充分利用多模态信息。
联合图像组学生物标志物的开发,以及从多模态模型中识别可解释的形态和分子描述符,对改善癌症预后评估至关重要。
1-2:研究方法
- 数据收集:从TCGA获取14种癌症类型的5720例患者的6592张H&E全切片图像(WSIs)及对应的分子和临床数据,对数据进行严格筛选和预处理,确保数据质量和可用性。
- 多模态融合算法(MMF):构建包含注意力机制的多实例学习网络(AMIL)处理WSIs、自归一化网络(SNN)处理分子数据特征,以及采用克罗内克积融合的多模态融合层的模型架构。通过弱监督学习,融合两种模态数据预测癌症死亡风险,并解释各特征与高低风险患者的相关性。
- 模型训练与评估:对每种癌症类型进行5折交叉验证训练MMF模型,并与单模态的AMIL和SNN模型对比,使用交叉验证一致性指数(c-Index)、动态曲线下面积(生存AUC)、Kaplan-Meier曲线和对数秩检验评估模型性能。
- 模型可解释性分析:应用注意力和梯度基的可解释性方法,通过定制可视化工具展示WSIs的高分辨率注意力热图,利用Shapley加性解释(SHAP)风格的归因决策图可视化分子特征的归因权重和方向,构建PORPOISE平台展示结果。
1-3:研究结果
- 模型性能:MMF模型总体c-Index达到0.644,生存AUC为0.662,在12/14种癌症类型上c-Index最高,在多数癌症类型的患者风险分层上表现优于单模态模型。如在KIRP中,MMF的c-Index达0.816,相比单模态模型有显著提升。
- 特征分析:高风险患者的WSIs高关注区域与肿瘤细胞增多、肿瘤侵袭相关;低风险患者的高关注区域免疫细胞更多、肿瘤分级更低。在分子特征方面,确定了多种癌症类型的关键致癌基因和免疫相关基因,如LGG中的IDH1突变、BRCA中的PIK3CA突变等。
- 免疫细胞与预后关系:在9/14种癌症类型中,低风险患者高关注区域的TIL存在显著增加,表明TIL可作为预测癌症预后的形态学特征。
1-4:研究结论
多模态整合对多数癌症类型的患者风险分层有益,但部分癌症单模态算法也能达到类似效果,应根据癌症类型选择合适的模态。
PORPOISE平台使模型更透明、可解释,有助于临床医生和研究人员深入探究。
未来可通过优化模型、利用单细胞测序等技术提升研究水平,为癌症治疗决策提供更有力的支持。
二、PORPOISE工作流程
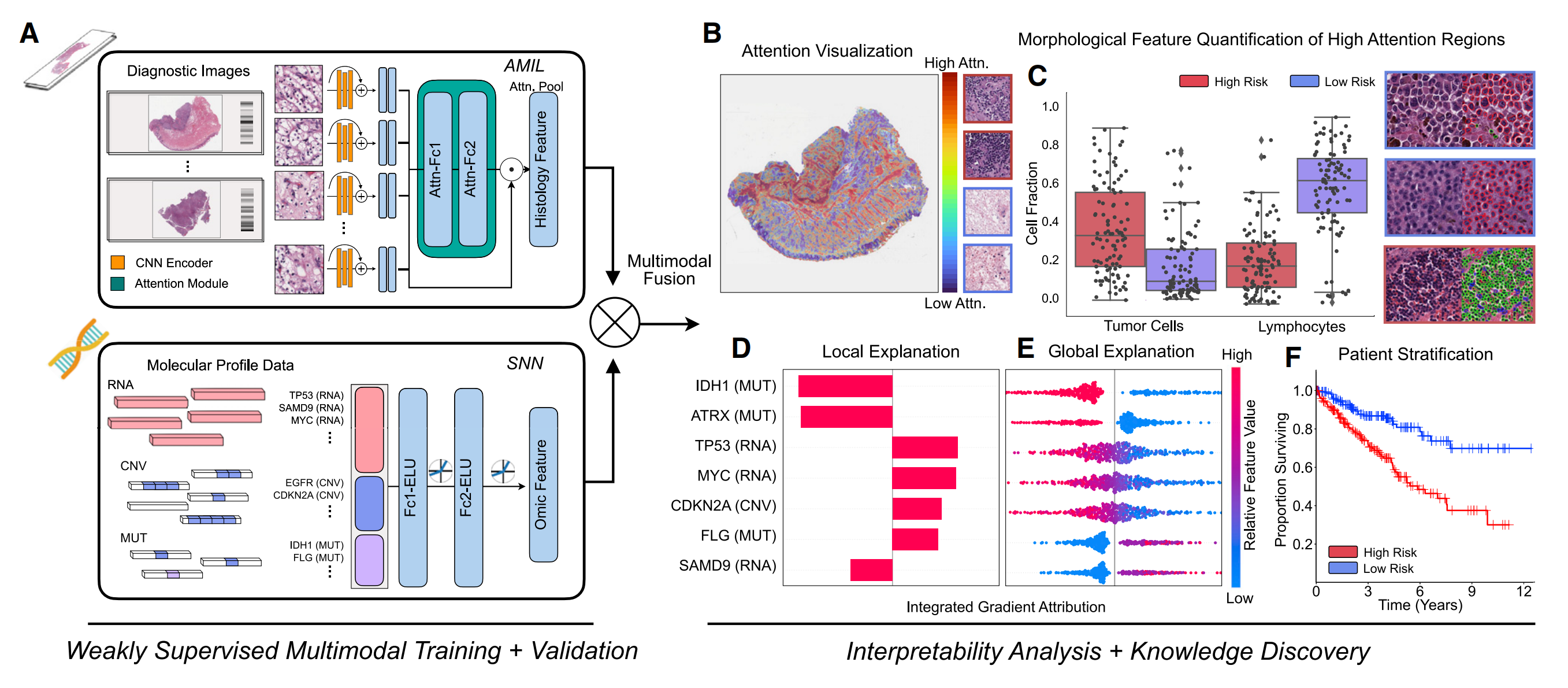
下图展示了PORPOISE(用于整合生存估计的病理 - 组学研究平台)的工作流程,主要分为“弱监督多模态训练与验证”和“可解释性分析与知识发现”两部分。
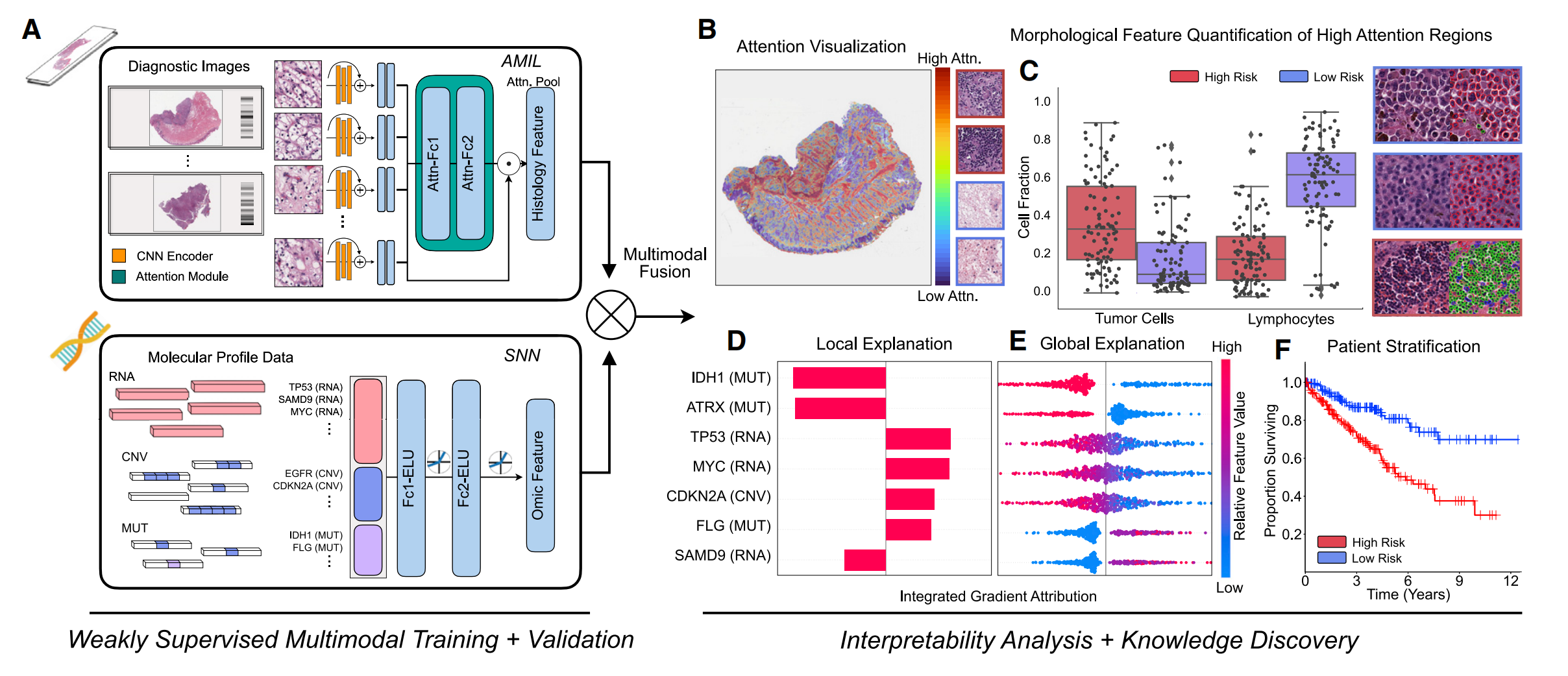
2-1:弱监督多模态训练与验证
- 输入数据:以数字化高分辨率福尔马林固定石蜡包埋(FFPE)的H&E组织学玻璃载片(即WSIs,全切片图像 )及对应的分子数据作为算法输入。
- 多模态算法模块
- AMIL(基于注意力的多实例学习网络 ):处理WSIs,通过CNN编码器提取图像特征,再经注意力模块得到组织学特征。
- SNN(自归一化网络 ):处理分子数据特征,如RNA、CNV(拷贝数变异)、MUT(突变)等相关数据。
- 多模态融合层:通过克罗内克积运算融合来自AMIL的组织学特征和SNN的组学特征 。
2-2:可解释性分析与知识发现
针对WSIs的分析
- 注意力可视化(B):利用基于注意力的可解释性方法,将每位患者的局部解释可视化为高分辨率注意力热图,热图中高关注区域(红色)对应对模型预测风险分数有贡献的形态学特征。
- 形态特征量化(C):提取高关注区域的形态学模式,对高风险和低风险患者队列的细胞比例(如肿瘤细胞、淋巴细胞)等进行量化分析 。
针对分子特征的分析
- 局部解释(D):使用基于归因的可解释性方法(整合梯度归因 ),将每位患者的分子特征局部解释可视化。
- 全局解释(E):通过分析所有患者基因归因的方向性、特征值和幅度,对分子特征进行全局可解释性分析 。
患者分层(F)
通过Kaplan - Meier分析对单个癌症类型的低风险和高风险患者进行分层可视化 。
三、PORPOISE性能及可解释性分析举例
这张图是对PORPOISE在肾透明细胞癌(KIRC)上的定量性能、局部模型解释和全局可解释性分析。
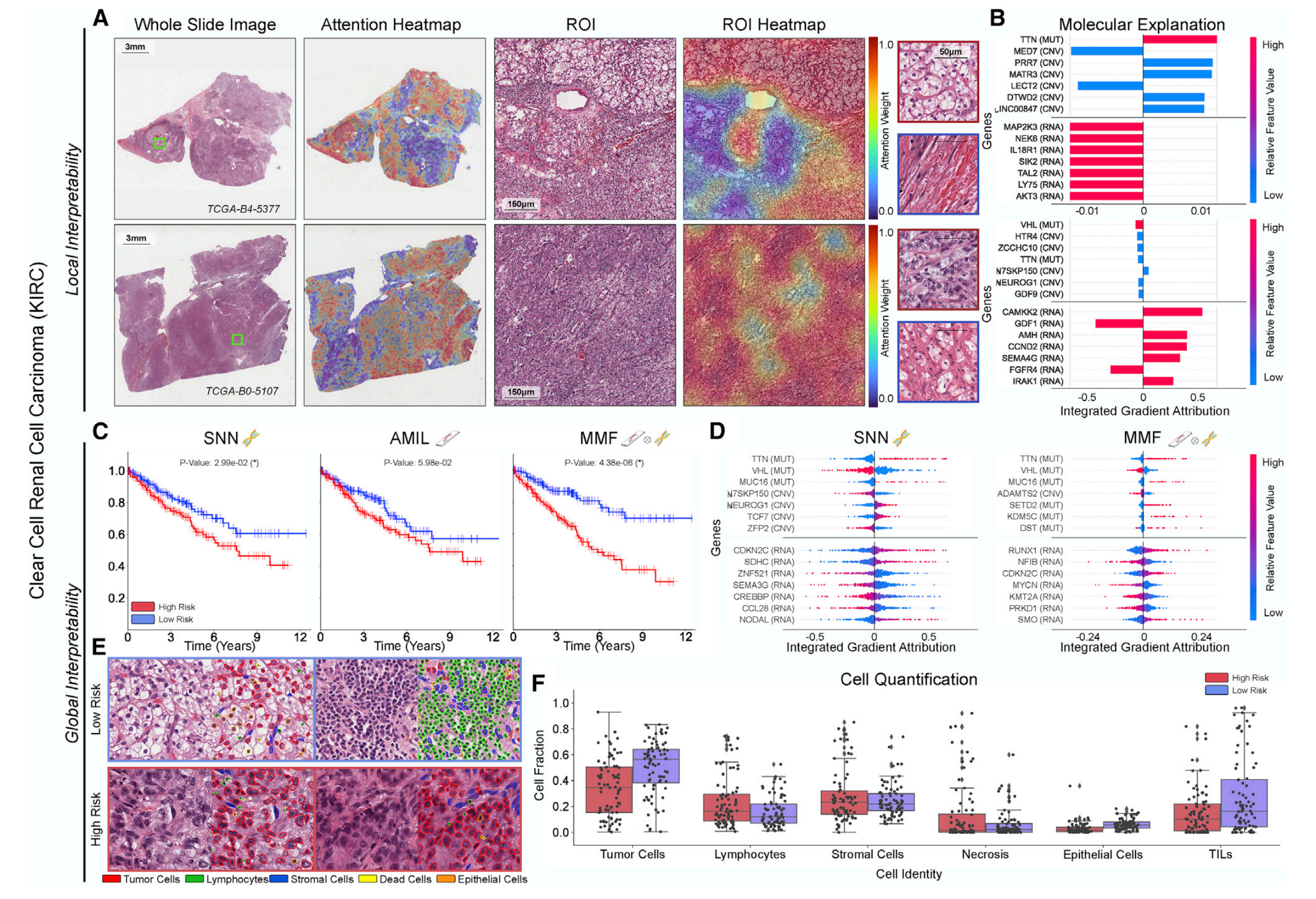
3-1:局部可解释性
- A. 图像可视化:呈现全切片图像(Whole Slide Image)、注意力热图(Attention Heatmap)、感兴趣区域(ROI)及其热图(ROI Heatmap)。不同颜色的注意力热图反映模型关注程度,低风险样本(如TCGA - 84 - S377 )关注区域细胞形态特征明显,高风险样本(如TCGA - 80 - 5107 )关注区域细胞形态有差异,可辅助理解模型关注的形态学特征。
- B. 分子解释:通过整合梯度归因展示分子特征对模型预测的贡献。不同基因以条形表示,颜色代表相对特征值高低,正负反映对高低风险预测的影响方向,帮助确定与KIRC预后相关的关键分子因素 。
3-2:全局可解释性
- C. Kaplan - Meier曲线:展示仅使用组学数据的SNN模型、仅使用组织学数据的AMIL模型和多模态融合的MMF模型的生存曲线。MMF模型曲线在高低风险患者分层上效果更好(p值更小 ),说明多模态融合在区分患者风险上优势显著。
- D. 全局基因归因:对比SNN和MMF模型对基因的全局归因情况。MMF能识别更多与免疫相关和预后相关基因(如RUNX1、NFEB等 ),拓展对KIRC分子机制与预后关联的认知。
- E. 细胞量化示例:呈现高低风险样本高关注区域的细胞类型分布图像。低风险区域淋巴细胞(绿色 )等免疫细胞丰富,高风险区域肿瘤细胞(红色 )占比大,直观显示细胞组成差异。
- F. 细胞量化:对高关注区域各类细胞(肿瘤细胞、淋巴细胞、基质细胞等 )进行量化统计。箱线图展示高低风险组细胞比例差异,如高风险组肿瘤细胞比例高,低风险组淋巴细胞和TILs(肿瘤浸润淋巴细胞 )比例高,为理解肿瘤微环境与预后关系提供量化依据。
四、方法
4-1:数据集描述
本文从癌症基因组图谱(TCGA)中14种癌症类型的5,720例患者处收集了6,592张苏木精 - 伊红(H&E)诊断全切片图像(WSIs)以及对应的分子和临床数据。
在数据集收集过程中,样本纳入标准依据以下两点确定:
- 1)每个TCGA项目中的数据集规模以及截尾与非截尾患者的分布情况;
- 2)每张WSI是否具备匹配的拷贝数变异(CNV)、突变和RNA测序丰度数据(WSI - CNV - MUT - RNA) 。
为避免在生存分析中对生存分布建模时出现过拟合,患者数量少于150例(在WSI和分子数据对齐后)且截尾情况不佳(非截尾患者比例低于5%)的TCGA项目被排除在研究之外。
补充:何为截尾情况不佳?
在生存分析的情境下,“截尾”指由于某些原因(如研究结束时患者仍存活、患者失访等 ),无法获取个体完整的生存时间信息。
“截尾情况不佳” 在这里是指在相关研究项目中,非截尾患者(即能够获取完整生存时间信息的患者 )占总患者数量的比例过低,具体到文中就是非截尾患者比例低于5% 。
这样低比例的非截尾患者,会使生存分析所依据的数据完整性不足,难以准确刻画生存分布,进而在对生存分布建模时容易出现过拟合问题,不利于可靠地分析患者生存情况及相关因素,所以这类项目被排除在研究之外。
对于胃肠道,这些器官的癌症类型被分别归为一组,形成合并的TCGA项目 - COADREAD(结肠癌(COAD)和直肠癌(READ)腺癌) 。
对于低级别胶质瘤(LGG),在模型训练时纳入了TCGA胶质瘤队列中的其他病例(如胶质母细胞瘤),但仅对LGG病例进行评估和可解释性分析 。
对于数据缺失较多的皮肤皮肤黑色素瘤(TCGA - SKCM)和子宫体子宫内膜癌(TCGA - UCEC),放宽了数据对齐标准,分别纳入仅匹配WSI - MUT - RNA和WSI - CNV - MUT的样本 。需注意,在TCGA - SKCM中,由于原发性肿瘤的匹配分子谱信息极少,因此也纳入了转移性病例 。
为降低分子谱数据中的特征稀疏性,使用了在每种癌症研究中CNV频率大于10%或突变频率大于5%的基因。在TCGA - SKCM和TCGA - UCEC中,使用10%的突变频率阈值,因为使用其他癌症类型的阈值会导致基因特征极少甚至为零 。
为限制RNA测序的特征数量,采用了来自分子特征数据库(Molecular Signatures Database)基因家族类别的基因集(Subramanian等人,2005) 。
分子和临床数据取自经过质量控制的cBioPortal文件。队列特征、截尾统计和特征对齐的汇总表见表格S1和S3。
4-2:WSI处理
对于每张WSI,利用用于WSI分析的公共CLAM资源库(Lu等人,2021)对组织进行自动分割 。
分割后,从所有识别出的组织区域,在20×等效金字塔层级提取大小为256×256且无重叠的图像块。随后,将在ImageNet上预训练的ResNet50模型用作编码器,在第3个残差块后通过空间平均池化,将每个256×256的图像块转换为1024维的特征向量。
为加快该过程,使用多个图形处理单元(GPU)并行计算,每个GPU的批量大小为128。
4-3:基于深度学习的整合全切片图像和基因组特征的生存分析
PORPOISE(用于整合生存估计的病理 - 组学研究平台)采用一种高通量、可解释、弱监督的多模态深度学习算法(MMF),旨在弱监督学习任务(如通过生存分析进行患者层面的癌症预后评估)中整合全切片图像和分子谱数据 。
给定:
- 1)作为金字塔文件的诊断WSIs;
- 2)单个患者经过处理的基因组和转录组特征,MMF学习联合表征这两种异质数据模态 。
尽管该算法用于生存分析,但可适用于任何模态组合,且能灵活解决计算病理学中具有患者层面标签的任何学习任务。
该算法由三个组件构成:
- 1)用于处理WSIs的基于注意力的多实例学习(AMIL);
- 2)用于处理分子谱数据的自归一化网络(SNN);
- 3)用于整合WSIs和分子谱数据的多模态融合层(由Pathomic Fusion扩展而来)(Chen等人,2020;Ilse等人,2018;Klambauer等人,2017;Lu等人,2021) 。
4-4:AMIL(基于注意力的多实例学习算法)
算法用途与背景
为从全切片图像(WSIs)进行生存预测,扩展了原本用于弱监督分类的基于注意力的多实例学习算法。
在多实例学习框架下,将超高像素的WSI划分为较小区域,把每个WSI视为图像块(实例)的集合(包) ,训练时使用对应的载片级别标签。
数据表示
WSI处理后,每个WSI包用 M i × C M_{i}\times C Mi×C矩阵张量表示 , M i M_{i} Mi是图像块数量(包大小,不同载片间有差异 ), C C C是特征维度(使用ResNet50编码器时 C = 1024 C = 1024 C=1024 )。
训练和评估时,将与同一患者病例对应的所有WSIs视为单个WSI包。
若患者有 N N N个WSI包,包大小分别为 M 1 , ⋯ , M N M_{1},\cdots,M_{N} M1,⋯,MN ,对应患者的WSI包由这 N N N个包连接而成,维度为 M × 1024 M\times1024 M×1024 ,其中 M = ∑ i = 1 N M i M=\sum_{i = 1}^{N}M_{i} M=∑i=1NMi。
模型组成
模型由投影层 f p f_{p} fp 、注意力模块 f a t t n f_{attn} fattn和预测层 f p r e d f_{pred} fpred组成。
每个WSI包的图像块嵌入 H ∈ R M × 1024 \mathbf{H}\in\mathbb{R}^{M\times1024} H∈RM×1024 ,先由全连接层 f p f_{p} fp(权重 W p r o j ∈ R 512 × 1024 \mathbf{W}_{proj}\in\mathbb{R}^{512\times1024} Wproj∈R512×1024 ,偏置 b b i a s ∈ R 512 \mathbf{b}_{bias}\in\mathbb{R}^{512} bbias∈R512 )映射到特定数据集的512维特征空间 。接着,注意力模块 f a t t n f_{attn} fattn为每个区域打分,衡量其与患者层面预后预测的相关性。
高注意力分数区域在聚合患者WSI所有区域信息时,对患者层面特征表示贡献更大(注意力池化操作 )。 f a t t n f_{attn} fattn由3个全连接层构成,权重分别为 U a ∈ R 256 × 512 \mathbf{U}_{a}\in\mathbb{R}^{256\times512} Ua∈R256×512 、 V a ∈ R 256 × 512 \mathbf{V}_{a}\in\mathbb{R}^{256\times512} Va∈R256×512和 W a ∈ R 1 × 256 \mathbf{W}_{a}\in\mathbb{R}^{1\times256} Wa∈R1×256 。
给定图像块嵌入 h m ∈ R 512 \mathbf{h}_{m}\in\mathbb{R}^{512} hm∈R512( H \mathbf{H} H的第 m m m行 ),其注意力分数 a m a_{m} am计算公式为:
a m = exp { W a ( tanh ( V a h m ⊤ ) ⊙ s i g m ( U a h m ⊤ ) ) } ∑ m = 1 M exp { W a ( tanh ( V a h m ⊤ ) ⊙ s i g m ( U a h m ⊤ ) ) } a_{m}=\frac{\exp\{\mathbf{W}_{a}(\tanh(\mathbf{V}_{a}\mathbf{h}_{m}^{\top})\odot\mathrm{sigm}(\mathbf{U}_{a}\mathbf{h}_{m}^{\top}))\}}{\sum_{m = 1}^{M}\exp\{\mathbf{W}_{a}(\tanh(\mathbf{V}_{a}\mathbf{h}_{m}^{\top})\odot\mathrm{sigm}(\mathbf{U}_{a}\mathbf{h}_{m}^{\top}))\}} am=∑m=1Mexp{Wa(tanh(Vahm⊤)⊙sigm(Uahm⊤))}exp{Wa(tanh(Vahm⊤)⊙sigm(Uahm⊤))}
注意力池化操作
注意力池化操作使用计算出的注意力分数(向量 A ∈ R M \mathbf{A}\in\mathbb{R}^{M} A∈RM )作为权重系数,将图像块层面的特征表示聚合为患者层面的特征表示 h p a t i e n t ∈ R 512 \mathbf{h}_{patient}\in\mathbb{R}^{512} hpatient∈R512 ,公式为:
h p a t i e n t = A t t n p o o l ( A , H ) = ∑ m = 1 M a m h m \mathbf{h}_{patient}=\mathrm{Attnpool}(\mathbf{A},\mathbf{H})=\sum_{m = 1}^{M}a_{m}\mathbf{h}_{m} hpatient=Attnpool(A,H)=m=1∑Mamhm
患者层面预测分数计算
最终的患者层面预测分数 s \mathbf{s} s ,通过预测层 f p r e d f_{pred} fpred(权重 W p r e d ∈ R 4 × 512 \mathbf{W}_{pred}\in\mathbb{R}^{4\times512} Wpred∈R4×512 ,采用sigmoid激活函数 )对包表示进行计算得到,即 s = f p r e d ( h b a g ) \mathbf{s}=f_{pred}(\mathbf{h}_{bag}) s=fpred(hbag) 。
最后一个全连接层用于学习表示 h W S I ∈ R 32 × 1 \mathbf{h}_{WSI}\in\mathbb{R}^{32\times1} hWSI∈R32×1 ,并作为多模态融合层的输入。
4-5:SNN(自归一化网络)用于分子特征生存预测
SNN背景
在使用分子特征进行生存预测时,采用自归一化网络(SNN) ,它已被证明在高维小样本(HDLSS)场景(如基因组数据从几百到几千个样本 )下效果良好(Klambauer等人,2017) 。
传统前馈网络在这种场景易过拟合,且当前深度学习正则化技术(如随机梯度下降和Dropout )存在训练不稳定问题。
SNN正则化技术
为在高维小样本基因组数据上使用更稳健的正则化技术,采用SNN架构中的归一化激活和Dropout层:
- 缩放指数线性单元(SeLU):与当前前馈网络中常见的修正线性单元(ReLU)激活函数相比,SeLU能使每层传播过程中输出趋向零均值和单位方差。其激活函数定义为 S e L U ( x ) = λ { x if x > 0 α e x − α if x ≤ 0 \mathrm{SeLU}(x)=\lambda\begin{cases}x & \text{if }x > 0\\\alpha e^{x}-\alpha & \text{if }x\leq0\end{cases} SeLU(x)=λ{xαex−αif x>0if x≤0 ,其中 α ≈ 1.67 \alpha\approx1.67 α≈1.67 , λ ≈ 1.05 \lambda\approx1.05 λ≈1.05 。
- Alpha Dropout:为在Dropout后保持归一化,对于给定层中的神经元,激活值不是以 1 − q 1 - q 1−q( 0 < q ≥ 1 0 < q\geq1 0<q≥1 )的概率设为0 ,而是设为 lim x → − ∞ S e L U ( x ) = − λ α = α ′ \lim_{x\rightarrow-\infty}\mathrm{SeLU}(x)=-\lambda\alpha=\alpha' limx→−∞SeLU(x)=−λα=α′ ,确保自归一化属性并更新均值和方差 ,相关公式为 E ( x d + α ′ ( 1 − d ) ) = q μ + ( 1 − q ) α ′ \mathbb{E}(xd+\alpha'(1 - d)) = q\mu+(1 - q)\alpha' E(xd+α′(1−d))=qμ+(1−q)α′ , V a r ( x d + α ′ ( 1 − d ) ) = q ( ( 1 − q ) ( α ′ − μ ) 2 + ν ) \mathrm{Var}(xd+\alpha'(1 - d)) = q((1 - q)(\alpha'-\mu)^{2}+\nu) Var(xd+α′(1−d))=q((1−q)(α′−μ)2+ν) 。
SNN架构
用于分子特征输入的SNN架构包含2个隐藏层,每层有256个神经元,每层都应用SeLU激活和Alpha Dropout 。
最后一个全连接层用于学习表示 h m o l e c u l a r ∈ R 32 × 1 \mathbf{h}_{molecular}\in\mathbb{R}^{32\times1} hmolecular∈R32×1 ,并作为多模态融合层的输入。
研究还通过去除自归一化和 L 1 L_{1} L1正则化的全连接层对MMF进行消融实验,发现自归一化和 L 1 L_{1} L1正则化对多模态训练很重要。
4-6:多模态融合层
构建多模态特征表示
在通过AMIL和SNN子网络构建单模态特征表示后,使用克罗内克积融合(Kronecker Product Fusion)学习多模态特征表示,以捕捉两种模态间的重要交互(Chen等人,2020;Zadeh等人,2017) 。
联合多模态张量通过 h W S I \mathbf{h}_{WSI} hWSI和 h m o l e c u l a r \mathbf{h}_{molecular} hmolecular的克罗内克积计算得到,即将 h m o l e c u l a r \mathbf{h}_{molecular} hmolecular中的每个神经元与 h W S I \mathbf{h}_{WSI} hWSI中的每个神经元相乘,以捕捉所有双模态交互。
为保留单模态特征,在融合前给每个单模态特征表示添加“1” ,公式为 h f u s i o n = [ h W S I 1 ] ⊗ [ h m o l e c u l a r 1 ] \mathbf{h}_{fusion} = \begin{bmatrix}\mathbf{h}_{WSI}\\1\end{bmatrix}\otimes\begin{bmatrix}\mathbf{h}_{molecular}\\1\end{bmatrix} hfusion=[hWSI1]⊗[hmolecular1] 。其中 ⊗ \otimes ⊗是克罗内克积, h f u s i o n ∈ R 33 × 33 \mathbf{h}_{fusion}\in\mathbb{R}^{33\times33} hfusion∈R33×33是可微的多模态张量,以 O ( 1 ) O(1) O(1)计算量对所有单模态和双模态交互进行建模。
基于门控的注意力机制
为减少单模态特征中的噪声影响并降低WSI与分子特征模态间的特征共线性,采用基于门控的注意力机制,额外控制每种模态的表达性 。
相关公式为:
h i , gated = z i ∗ h i , ∀ h i ∈ { h W S I , h m o l e c u l a r } \mathbf{h}_{i, \text{gated}} = \mathbf{z}_{i}*\mathbf{h}_{i}, \forall \mathbf{h}_{i}\in\{\mathbf{h}_{WSI}, \mathbf{h}_{molecular}\} hi,gated=zi∗hi,∀hi∈{hWSI,hmolecular}
h i = R e L U ( W j ⋅ h i ) \mathbf{h}_{i} = \mathrm{ReLU}(W_{j}\cdot\mathbf{h}_{i}) hi=ReLU(Wj⋅hi)
z i = σ ( W j ⋅ [ h W S I , h m o l e c u l a r ] ) \mathbf{z}_{i} = \sigma(W_{j}\cdot[\mathbf{h}_{WSI}, \mathbf{h}_{molecular}]) zi=σ(Wj⋅[hWSI,hmolecular])
对于学习到单模态特征 h i \mathbf{h}_{i} hi的模态 i i i ,学习权重矩阵 W j W_{j} Wj对模态 i i i中每个特征的相对重要性进行打分。
在进行Softmax操作后, z i \mathbf{z}_{i} zi可视为 h W S I \mathbf{h}_{WSI} hWSI和 h m o l e c u l a r \mathbf{h}_{molecular} hmolecular对 h i \mathbf{h}_{i} hi中每个特征的注意力分数。通过将原始单模态特征 h i \mathbf{h}_{i} hi与注意力分数 z i \mathbf{z}_{i} zi进行元素相乘得到门控表示 h i , gated \mathbf{h}_{i, \text{gated}} hi,gated 。
在基于门控的注意力实现中,在融合前对两种模态都应用门控,并对多模态网络的倒数第二层隐藏层添加额外的跳跃连接 。
后续处理
经过克罗内克积融合后,多模态张量通过两个大小为256的隐藏层进行传播,然后使用基于交叉熵的损失函数进行监督,以用于生存分析 。
表格S3展示了仅对病理特征进行门控、仅对基因组特征进行门控以及在克罗内克积融合前对病理和基因组特征都进行门控的消融实验研究 。
4-7:生存损失函数
数据离散化
为从右截尾的患者层面生存数据进行生存预测,首先将患者总体生存时间(以月为单位, T c o n t T_{cont} Tcont )的连续时间尺度划分为4个不重叠的区间 [ t 0 , t 1 ) [t_0, t_1) [t0,t1)、 [ t 1 , t 2 ) [t_1, t_2) [t1,t2)、 [ t 2 , t 3 ) [t_2, t_3) [t2,t3)、 [ t 3 , t 4 ) [t_3, t_4) [t3,t4) ,其中 t 0 = 0 t_0 = 0 t0=0 , t 4 = ∞ t_4 = \infty t4=∞ , t 1 t_1 t1、 t 2 t_2 t2、 t 3 t_3 t3定义为非截尾患者事件时间的四分位数 。
对于数据集中索引为 j j j、对应随访时间 T j , c o n t ∈ [ 0 , ∞ ) T_{j, cont}\in[0, \infty) Tj,cont∈[0,∞)的每个患者记录,当 T j , c o n t ∈ [ t r , t r + 1 ) T_{j, cont}\in[t_r, t_{r + 1}) Tj,cont∈[tr,tr+1)时,定义 T j = r T_j = r Tj=r 。将第 j j j个患者离散化的真实标签记为 Y j Y_j Yj 。
风险函数与生存函数建模
对于具有包层面表示 h b a g j \mathbf{h}_{bagj} hbagj的给定患者,预测层 f p r e d f_{pred} fpred(权重 W p r e d ∈ R 4 × 512 \mathbf{W}_{pred}\in\mathbb{R}^{4\times512} Wpred∈R4×512 )对风险函数建模为 f h a z a r d ( r ∣ h b a g j ) = P ( T j = r ∣ T j ≥ r , h b a g j ) f_{hazard}(r|\mathbf{h}_{bagj}) = P(T_j = r|T_j\geq r, \mathbf{h}_{bagj}) fhazard(r∣hbagj)=P(Tj=r∣Tj≥r,hbagj) 。
风险函数与生存函数的关系为 f s u r v ( r ∣ h b a g j ) = P ( T j > r ∣ h b a g j ) = ∏ u = 1 r ( 1 − f h a z a r d ( u ∣ h b a g j ) ) f_{surv}(r|\mathbf{h}_{bagj}) = P(T_j > r|\mathbf{h}_{bagj})=\prod_{u = 1}^{r}(1 - f_{hazard}(u|\mathbf{h}_{bagj})) fsurv(r∣hbagj)=P(Tj>r∣hbagj)=∏u=1r(1−fhazard(u∣hbagj)) 。
对数似然损失函数
为优化模型参数,使用离散生存模型的对数似然函数(Zadeh和Schmid,2020) 。给定二元截尾状态 c j c_j cj ,损失函数 L L L表示为:
L = − c j ⋅ log ( f s u r v ( Y j ∣ h b a g j ) ) − ( 1 − c j ) ⋅ log ( f s u r v ( Y j − 1 ∣ h b a g j ) ) − ( 1 − c j ) ⋅ log ( f h a z a r d ( Y j ∣ h b a g j ) ) \begin{align*} L&=-c_j\cdot\log(f_{surv}(Y_j|\mathbf{h}_{bagj}))-(1 - c_j)\cdot\log(f_{surv}(Y_j - 1|\mathbf{h}_{bagj}))-(1 - c_j)\cdot\log(f_{hazard}(Y_j|\mathbf{h}_{bagj})) \end{align*} L=−cj⋅log(fsurv(Yj∣hbagj))−(1−cj)⋅log(fsurv(Yj−1∣hbagj))−(1−cj)⋅log(fhazard(Yj∣hbagj))
其中,随访期结束后仍存活的患者 c j = 1 c_j = 1 cj=1 ,在 T j , c o n t T_{j, cont} Tj,cont时刻死亡的患者 c j = 0 c_j = 0 cj=0 。
训练时,通过最小化 L L L与 L u n c e n s o r e d L_{uncensored} Luncensored的加权和来强调非截尾患者病例的贡献
L s u r v = ( 1 − β ) ⋅ L + β ⋅ L u n c e n s o r e d L_{surv}=(1 - \beta)\cdot L+\beta\cdot L_{uncensored} Lsurv=(1−β)⋅L+β⋅Luncensored
对应非截尾患者的损失函数第二项 L u n c e n s o r e d L_{uncensored} Luncensored定义为:
L u n c e n s o r e d = − ( 1 − c j ) ⋅ log ( f s u r v ( Y j − 1 ∣ h b a g j ) ) − ( 1 − c j ) ⋅ log ( f h a z a r d ( Y j ∣ h b a g j ) ) \begin{align*} L_{uncensored}&=-(1 - c_j)\cdot\log(f_{surv}(Y_j - 1|\mathbf{h}_{bagj}))-(1 - c_j)\cdot\log(f_{hazard}(Y_j|\mathbf{h}_{bagj})) \end{align*} Luncensored=−(1−cj)⋅log(fsurv(Yj−1∣hbagj))−(1−cj)⋅log(fhazard(Yj∣hbagj))
4-8:训练细节
数据集划分
对于每种疾病模型研究,患者病例被随机划分为不重叠的训练集(80%)和测试集(20%) ,用于训练模型和评估性能。
划分在患者病例层面进行,即同一患者病例的所有切片仅出现在测试集或训练集中,不会同时用于训练和测试 。
交叉验证
对每种疾病模型进行五次重复实验,采用五折交叉验证,五次重新分配患者病例到不重叠的训练和测试队列中 。
训练和评估MMF(多模态融合模型)及单模态模型都采用相同流程 。
训练参数
对于所有癌症类型,MMF模型使用Adam优化器进行端到端训练,涉及AMIL子网络、SNN子网络和多模态融合层。
学习率为 2 × 1 0 − 4 2\times10^{-4} 2×10−4 , b 1 b_1 b1系数为 0.9 0.9 0.9 , b 2 b_2 b2系数为 0.999 0.999 0.999 , L 2 L_2 L2权重衰减为 1 × 1 0 − 5 1\times10^{-5} 1×10−5 , L 1 L_1 L1权重衰减为 1 × 1 0 − 5 1\times10^{-5} 1×10−5 ,训练轮数为20轮 。由于患者样本的WSIs图像尺寸各异,以1为小批量大小随机采样成对的WSIs和分子特征 。
在与单模态网络进行比较分析时,AMIL和SNN也使用与MMF相同的生存损失函数和超参数独立训练 。
4-9:计算硬件与软件
使用Python 3.7.7 ,涉及的软件包包括PyTorch 1.3.0 、Lifelines 0.24.6 、NumPy 1.18.1 、Pandas 1.1.3 、PIL 7.0.0 、OpenSlide 1.1.1 。
所有WSIs在Intel Xeon多核CPU及16个2080 Ti GPU上处理,借助自定义且公开可用的CLAM全切片处理流程 。多模态融合层通过自定义公开的Pathomic Fusion软件实现。
深度学习模型使用Nvidia的CUDA 11.0和cuDNN 7.5训练。Integrated Gradients通过Captum 0.2.0实现。细胞实例分割与分类借助HoVerNet软件。
统计分析(双样本t检验、对数秩检验)分别使用SciPy 1.4.1和Lifelines 0.24.6 。绘图与可视化使用Seaborn 0.9.0 、Matplotlib 3.1.1和Shap 0.35.0 。
4-10:量化与统计分析
生存曲线绘制与检验
绘制Kaplan - Meier曲线时,汇总验证集中的样本外风险预测结果并对应生存时间绘制 。
在Kaplan - Meier分析中对患者分层进行显著性检验,使用对数秩检验判断两种生存分布差异是否有统计学意义(P值<0.05 ) 。
模型性能评估指标
- c - Index:交叉验证的c - Index性能以5折平均c - Index报告。通过对验证集中样本外预测进行1000次非参数自助法估计95%置信区间 。
- 生存AUC:除c - Index外,还报告累积/动态AUC(生存AUC ),衡量模型在不同时间点对患者风险分层能力,并通过计算截尾加权逆概率校正截尾带来的乐观偏差 。
特征显著性评估
- 形态特征:对高低风险患者前1%高关注区域的细胞类型平均比例分布进行双样本t检验,评估单个细胞类型存在的全局形态特征显著性(P值<0.05 ) 。
- 分子特征:对高低基因特征值(分别为基因特征值中位数上下 )的归因分布进行双样本t检验,评估单个基因特征的全局分子特征显著性 。箱线图中,箱体表示数据分布的第1、中位数和第3四分位数,须线延伸至四分位距1.5倍范围内的数据点 。
结束语
本期推文的内容就到这里啦,如果需要获取医学AI领域的最新发展动态,请关注小罗的推送!如需进一步深入研究,获取相关资料,欢迎加入我的知识星球!