【数据分析】比较SparCC、Pearson和Spearman相关性估计方法在合成组学数据上的表现
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!
文章目录
-
- 介绍
- 加载R包
- 模拟数据
- 构建网络
- RMSE指数计算
- 画图
- 总结
- 系统信息
介绍
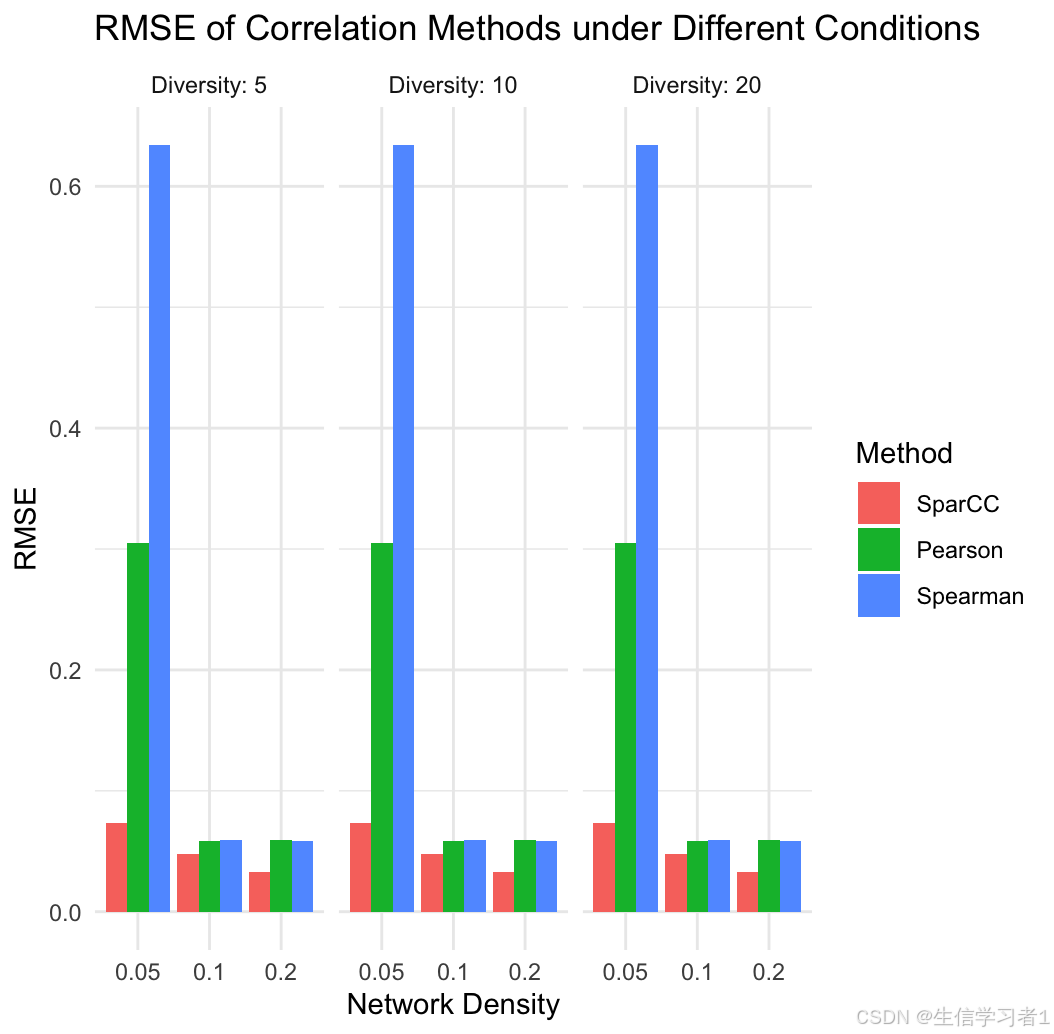
在生物信息学和生态学研究中,组学数据的分析越来越依赖于对微生物群落或基因表达数据中物种或基因间相关性的准确估计。传统的相关性估计方法,如Pearson和Spearman相关系数,虽然在处理连续数据时表现良好,但在处理组成数据时可能会遇到挑战。组成数据是由比例构成的,其总和固定,这使得数据的分布特性与传统的正态分布假设不符,从而影响相关性估计的准确性。
近年来,一种新的相关性估计方法——SparCC(Sparse Correlations for Compositional data)被提出,专门用于处理组成数据。SparCC方法通过稀疏表示和正则化技术,能够在控制假阳性率的同时,准确地估计组成数据中的相关性。然而,SparCC方法在不同数据特性(如多样性水平和网络密度)下的表现如何,以及与传统方法相比的优势和局限性,仍需进一步研究。
本研究通过模拟不同多样性水平和网络密度下的组成数据,比较了SparCC、Pearson和Spearman三种相关性估计方法的表现。首先,我们生成了合成的组成数据,模拟了不同多样性水平(5, 10, 20)和网络密度(0.05, 0.1, 0.2)条件下的微生物群落数据。然后,使用SparCC、Pearson和Spearman方法估计这些数据的相关性,并计算每种方法估计的相关性与真实相关性