100个GEO基因表达芯片或转录组数据处理27 GSE83456
写在前边
虽然现在是高通量测序的时代,但是GEO、ArrayExpress等数据库储存并公开大量的基因表达芯片数据,还是会有大量的需求去处理芯片数据,并且建模或验证自己所研究基因的表达情况,芯片数据的处理也可能是大部分刚学生信的道友入门R语言数据处理的第一次实战,因此准备更新100个基因表达芯片或转录组高通量数据的处理。
数据信息检索
可以看到GSE83456是基因表达芯片数据,因此可以使用GEOquery包 处理

使用GEOquery包下载数据
安装所需R包
BiocManager::install("lumi")
BiocManager::install("lumiHumanIDMapping")
BiocManager::install('ScienceAdvances/Canton')
注:using作用是一次性加载多个R包,不用写双引号,并且不在屏幕上打印包的加载信息
Canton::using(using, tidyverse,lumi,lumiHumanIDMapping, GEOquery, magrittr, data.table, AnnoProbe, clusterProfiler, org.Hs.eg.db, org.Mm.eg.db,ggdendro,ComplexHeatmap)
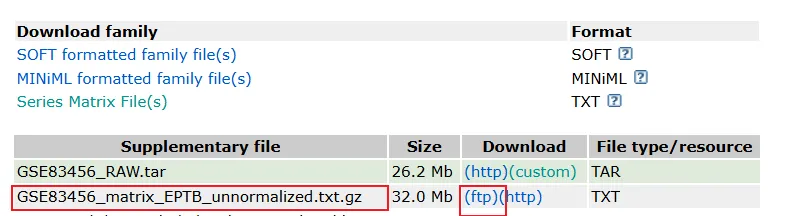
因为文件太大,在R内下载失败,可通过图片中的方法下载文件,GEOquery::getGEO直接读取本地的文件。
geo_accession <- "GSE83456"
eSet <- getGEO(filename=stringr::str_glue('{geo_accession}_series_matrix.txt.gz'), AnnotGPL = F, getGPL = F)


处理表型数据
这部分是很关键的,可以筛选一下分组表型信息,只保留自己需要的样本,作为后续分析的样本(根据自己的研究目的筛选符合要求的样本)
pdata <- pData(eSet)
pdata %<>%dplyr::mutate(Sample = geo_accession,Group = case_when(`disease state:ch1`=='HC'~'HC',`disease state:ch1`=='PTB'~'TB',TRUE~NA),Race = `ethnicity:ch1`,Sex = `gender:ch1`) %>%dplyr::select(Sample,Group,Race,Sex,everything())
处理表达谱数据
这里需要处理Illumina表达芯片的原始数据,因为GSE98895_series_matrix.txt.gz 中的基因表达值从-20到几千,无法用于后续分析,因此我们自己标准化处理.
下载原始数据
lumi代码主要两个步骤,读入数据和lumiExpresso(包括lumiB调整芯片背景;lumiT 对数据进行方差固定;lumiN 对方差固定后的数据进行标准化,lumiQ 评估数据质量。所有这些方法构成了一个预处理流程),可以参考lumi包发表的文章 PMID: 18467348

读入原始数据,并重命名行名,40个样本这里需要根据上面代码返回结果自己改动,前两行是探针id和对应的symbol,后面每两列对应一个样本,是荧光信号强度和pvalue
最后保存原始数据到tmp.txt文件中(参考生信技能树代码)
a <- fread('GSE83456_matrix_EPTB_unnormalized.txt.gz',data.table=F)colnames(a) <- c("PROBE_ID", paste(rep(rownames(pdata), each = 2),rep(c("AVG_Signal", "Detection Pval"), nrow(pdata)/2),sep = "."
))
a %<>% mutate(Gene=PROBE_ID,.after="PROBE_ID")
fwrite(a, file = "tmp.txt", sep = "\t", quote = F)探针信息
gpl=eSet$annotation
probe2symbol <- AnnoProbe::idmap(gpl = gpl, type = "bioc", mirror = "tencent", destdir = "./") %>%dplyr::rename(ProbeID = probe_id, Feature = symbol) # pipe", "bioc", "soft"
lumi处理原始数据并提取标准化后的表达矩阵
conflicts_prefer(data.table::second)
conflicts_prefer(dplyr::first)
x.lumi <- lumi::lumiR("tmp.txt", lib.mapping = "lumiHumanIDMapping")# BiocManager::install("preprocessCore", configure.args="--disable-threading", force = TRUE)
lumi.N.Q <- lumi::lumiExpresso(x.lumi)dat <- exprs(lumi.N.Q)
ID转换
第一次ID转换,把表达矩阵中的第一列ID转换成第二列PROBE_ID
probeid <- lumi.N.Q@featureData@data
probeid <- probeid[match(rownames(dat), rownames(probeid)),,drop=FALSE ]
identical(rownames(dat), rownames(probeid))
probeid$PROBE_ID -> rownames(dat)
| PROBE_ID | |
|---|---|
| rp13_p1x6D80lNLk3c | ILMN_1725881 |
| NEX0oqCV8.er4HVfU4 | ILMN_1910180 |
| KyqQynMZxJcruyylEU | ILMN_1804174 |
| xXl7eXuF7sbPEp.KFI | ILMN_1796063 |
| iKjLBHp0iJBJUEuiiU | ILMN_3284771 |
| 9xddNLgNfLqLuukhvY | ILMN_3208259 |
第二次ID转换,把ProbeID转换成基因名字,把表达矩阵中的探针名转换为基因名;transid是我写的一个R函数,有需要可以联系我,加入交流群
fdata <- transid(probe2symbol, probe_exprs)
| ProbeID | Feature |
|---|---|
| ILMN_1343291 | EEF1A1 |
| ILMN_1343295 | GAPDH |
| ILMN_1651209 | SLC35E2A |
| ILMN_1651221 | EFCAB1 |
| ILMN_1651228 | RPS28 |
| ILMN_1651229 | IPO13 |
数据质控
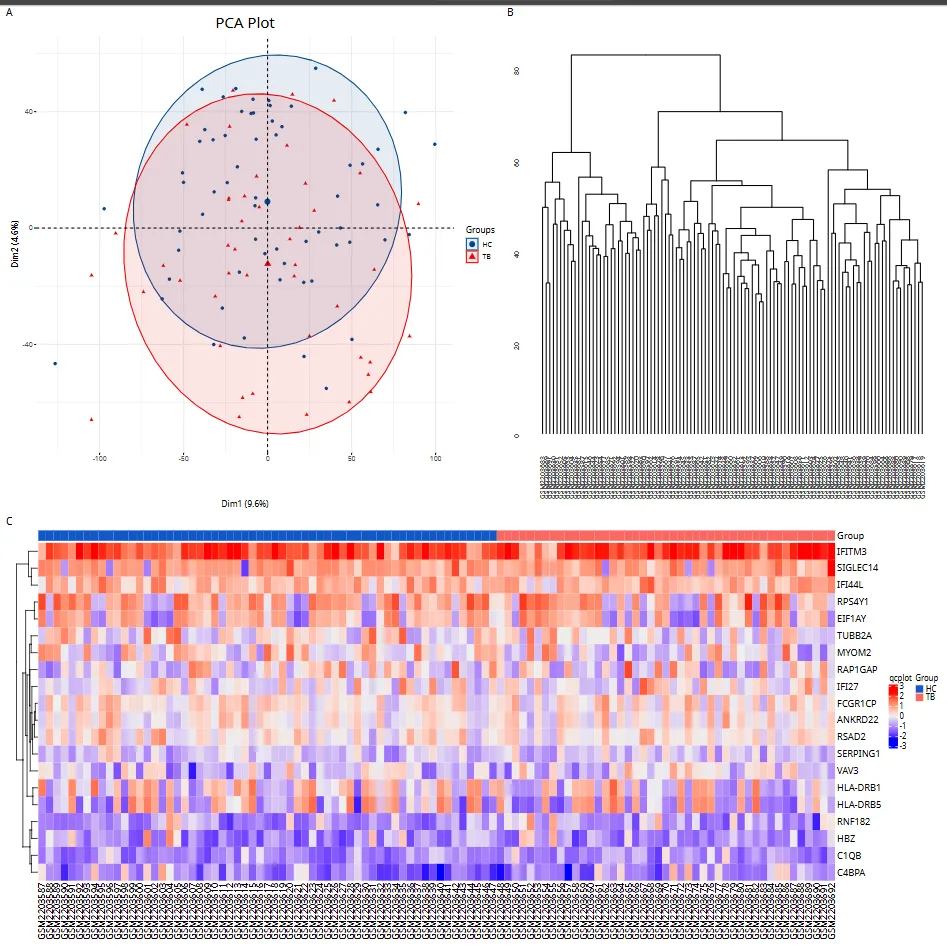
qcplot为自定义函数,作用是绘制用于质控判断的图,如PCA、top20基因热图、树状图,PCA图,可以 2 组区别区别不明显,可以考虑直接用作者处理好的数据虽然 有 负数值
common_samples <- base::intersect(colnames(fdata),pdata$Sample)
fdata %<>% dplyr::select(all_of(c("Feature",common_samples)))
pdata %<>% dplyr::filter(Sample %in% common_samples)qcplot(column_to_rownames(fdata,"Feature"),pdata$Group,w=18,h=18)
保存数据
fwrite(fdata, file = stringr::str_glue("{geo_accession}_{gpl}_fdata.csv.gz"))
fwrite(pdata, file = stringr::str_glue("{geo_accession}_{gpl}_pdata.csv"))