完整进行一次共线性分析
(随便找个基因家族)
1.数据收集
使用水稻、拟南芥、玉米三种作物进行示例
可以直接去ensemble去找最标准的基因组fasta文件和gff文件。
2.预处理数据
这里对于fasta和gff数据看情况要不要过滤掉线粒体叶绿体的基因,数据差异非常大,这里要自己写脚本处理一下
不同脚本:.....
import pandas as pdf_gff = pd.read_csv("Arabidopsis_thaliana.TAIR10.61.v3.gff3", sep=" ", comment="#", header=None)#不保留叶绿体 线粒体基因
f_gff = f_gff[f_gff[0].apply(lambda x: isinstance(x, int))]
f_gff[0] = f_gff[0].astype(str)f_gff[0] = 'Chr' + f_gff[0]#加上Chr前缀
f_gff.to_csv("Arabidopsis_thaliana.TAIR10.61.v4.gff3", sep=" ", header=None, index=False)
import pandas as pdf_gff = pd.read_csv("osa1_r7.all_models.v3.gff3", sep=" ", comment="#", header=None)
f_gff = f_gff[f_gff[0].str.match(r'Chr\d+')]
f_gff.to_csv("osa1_r7.all_models.v4.gff3", sep=" ", header=None, index=False)
然后:
#依次对数据进行预处理> 此处为linux虚拟机 conda环境下
——————————————————————————————————————————————————————————————————————
1.先对gff进行预处理,这里先用tbtools做了修复(gxf fix),然后这里再uniq去除一下冗余转录本:
python -m jcvi.formats.gff uniq --type=mRNA data/Arabidopsis/Arabidopsis_thaliana.TAIR10.61.v2.gff3 -o data/Arabidopsis/Arabidopsis_thaliana.TAIR10.61.v3.gff3 python -m jcvi.formats.gff uniq --type=mRNA data/maize/b73_fixed.gff3 -o data/maize/b73_v3.gff3 python -m jcvi.formats.gff uniq --type=mRNA data/rice/osa1_r7.all_models2.gff3 -o data/rice/osa1_r7.all_models.v3.gff3 ————————————————————————————————————————————————————————————————————————
#然后转bed 为了保险再primary_only一下、这里的指令肯定是要自己调整的,比如我的玉米gff里面gene没有Name字段,就要改成IDpython -m jcvi.formats.gff bed --type=mRNA --key=transcript_id --primary_only data/Arabidopsis/Arabidopsis_thaliana.TAIR10.61.v3.gff3 -o data/Arabidopsis/Arabidopsis_thaliana.TAIR10.61.bedpython -m jcvi.formats.gff bed --type=gene --key=ID --primary_only data/maize/b73_v2.gff3 -o data/maize/b73.bedpython -m jcvi.formats.gff bed --type=gene --key=Name --primary_only data/rice/osa1_r7.all_models.v3.gff3 -o data/rice/osa1_r7.all_models.bed——————————————————————————————————————————————————————————————————————————
到这里就获得了三个物种的bed文件
#最后一步要准备所有物种的cds,可以从官网下载,但是如果gff和fasta都没有问题了,也可以用tbtools直接提取,好像更不容易出错
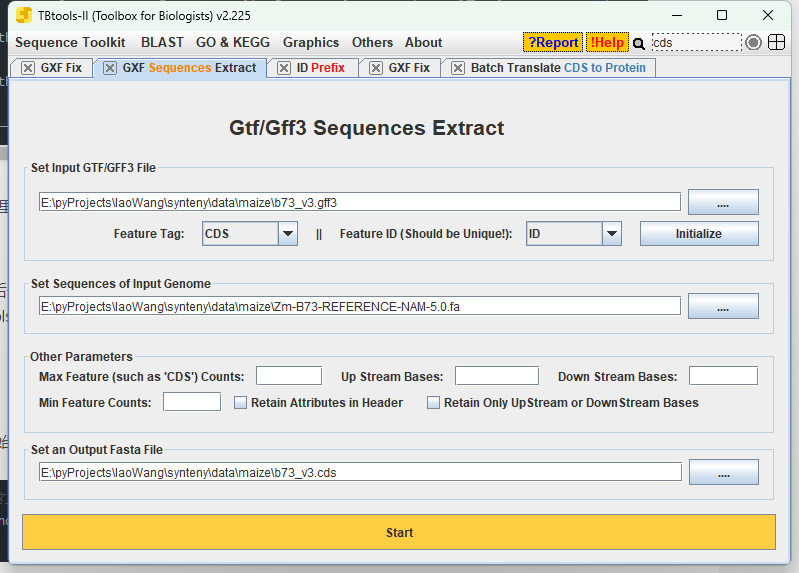
总之 你最后要整理这些文件:

并保持cds和bed中的名称一致
3.开始
#这里需要依赖
conda install bioconda::last先尝试两个物种间的情况#分别计算
#太严格基本匹配不到
python -m jcvi.compara.catalog ortholog maize Arabidopsis --cscore=.99 --no_strip_names#先使用这个把
python -m jcvi.compara.catalog ortholog maize Arabidopsis --no_strip_names python -m jcvi.compara.catalog ortholog maize rice --no_strip_names --exclude EXCLUDE这里生成.simple文件
python -m jcvi.compara.synteny screen --minspan=30 --simple maize.Arabidopsis.anchors maize.Arabidopsis.anchors.new python -m jcvi.compara.synteny screen --minspan=30 --simple maize.rice.anchors maize.rice.anchors.new 然后这里最终需要的是3个文件:
1.layout_2_1:
# y, xstart, xend, rotation, color, label, va, bed.6, .1, .8, 0, , maize, top, maize.bed.4, .1, .8, 0, , Arabidopsis, top, Arabidopsis.bed # edges e, 0, 1, maize.Arabidopsis.anchors.simple
2.seqids_2_1
chr1,chr2,chr3,chr4,chr5,chr6,chr7,chr8,chr9,chr10 1,2,3,4,5
3.maize.Arabidopsis.anchors.simple
python -m jcvi.graphics.karyotype seqids_2_1 layout_2_1python -m jcvi.graphics.karyotype --basepair seqids_2_3 layout_2_3还没写完
——————