水稻调控组全景的综合绘制与建模揭示了复杂性状背后的调控架构
摘要:
解析调控复杂性状的分子机制是推动作物改良的关键。本研究构建了水稻(Oryza sativa)迄今最全面的调控组图谱,系统描绘了3个代表性品种23种不同组织的染色质可及性景观。我们鉴定出117,176个独特的开放染色质区域(OCRs),占水稻基因组的约15%,该比例显著高于以往植物领域的报道。通过整合配对组织的RNA-seq数据,我们高置信度预测了59,075个OCR-基因调控关联,其中69.54%由增强子介导,并涵盖多个已知的增强子-基因互作案例。
基于该资源,我们重新评估了全基因组关联研究(GWAS)结果,发现转录因子OsbZIP06在种子萌发中的未知功能,并通过实验予以证实。我们优化深度学习模型以解析调控序列语法,实现了组织特异性染色质可及性的稳健建模。该方法可仅依据基因组序列预测品种间调控动态差异,为阐释顺式调控变异与品种形态分化的遗传基础提供了新视角。
综上,本研究为水稻功能基因组学与精准分子育种奠定了重要基础,并为解析复杂性状的调控机制提供了宝贵资源。
水稻(Oryza sativa)不仅是全球最重要的粮食作物之一,也是研究植物生长发育的卓越模式物种。过去二十年来,科研界投入巨大努力,以解析水稻重要农艺性状的遗传基础。其中,全基因组关联分析(GWAS)在这一进程中发挥了关键作用,成功将遗传变异与表型多样性联系起来。这些研究鉴定出大量有望用于性状改良的候选基因。然而,尽管取得了上述进展,我们对调控水稻复杂性状的分子机制仍缺乏完整认识。
基因调控网络(GRNs)在很大程度上由顺式调控 DNA 序列(如启动子和增强子)决定,这些序列可被特定的转录因子(TFs)识别并结合。解析这些调控序列内在的“调控密码”,并将调控序列与其靶基因准确关联,是重编程 GRNs、实现作物改良与性状优化的关键。然而,在水稻中,对“调控组”(regulome,即全基因组范围内所有调控元件的集合)的系统性刻画仍十分有限。现有研究往往聚焦于单一或少数组织,忽视了发育阶段和组织类型的全景式覆盖。同样,将调控区域与其靶基因建立可靠连接的努力亦显不足。
与此同时,许多与水稻农艺性状相关的功能遗传变异位于非编码调控区(例如 qSH1、DROT1 和 FZP),这使得它们的生物学解释极具挑战性,也凸显了对调控序列进行系统性解析的迫切需求。鉴于不同性状在特定发育阶段和组织中才显现出来,目前尚缺乏涵盖多种组织与生长时期的综合表观基因组图谱,这极大阻碍了水稻非编码调控变异的系统注释工作。
为弥合上述研究空白,我们采用实验室自主改良的 UMI-ATAC-seq 技术(一种优化的 ATAC-seq 方法),系统绘制了 3 个代表性水稻品种全生命周期多种组织的染色质可及性图谱。通过对 145 份 ATAC-seq 数据的整合分析,共鉴定出 117,176 个独特的开放染色质区域(OCRs),约占水稻基因组的 15%。结合配对组织的 RNA-seq 数据,我们基于跨组织的基因表达与邻近染色质可及性的相关性,预测了各 OCR 的潜在靶基因。
借助转录因子足迹(TF footprinting)分析,我们推断了组织或发育阶段特异性的调控网络;并通过比较籼稻与粳稻亚种的调控景观,鉴定出品种间多态/性状关联的 OCR。尤为重要的是,我们发现 GWAS 关联变异显著富集于组织特异性 OCR 中,依托这一 OCR 图谱,成功将 209 个复杂性状与非编码调控变异建立了因果关联。
进一步地,我们利用优化的深度学习模型,通过建模组织特异性染色质可及性并基于序列进行跨品种预测,解码了调控“语法”,揭示了导致顺式调控分化的关键遗传变异。综上,这些数据不仅成为植物研究领域的基石资源,也为精准分子育种提供了宝贵的调控变异位点。
结果
绘制水稻染色质可及性参考图谱
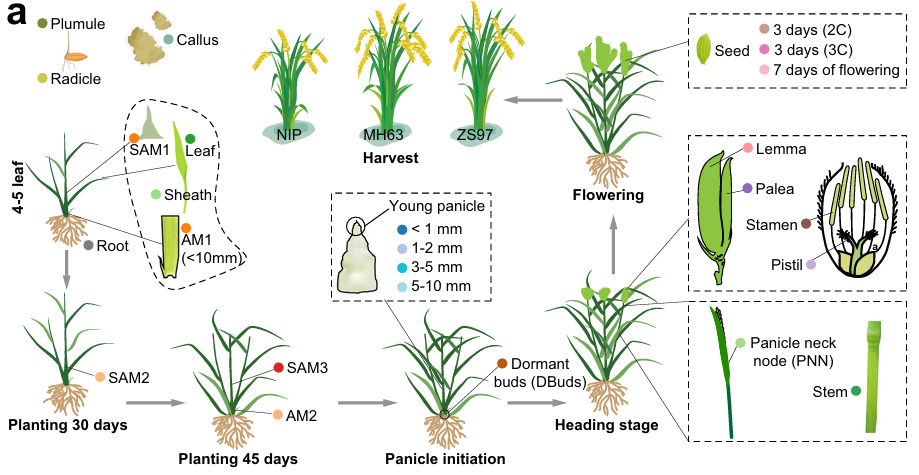
为了全面描绘水稻(Oryza sativa)开放染色质的景观,我们采用改进的 ATAC-seq 方法——UMI-ATAC-seq(在常规 ATAC-seq 基础上引入唯一分子标签,实现精确定量与足迹分析),对覆盖水稻整个生命周期的 23 种组织/器官进行染色质可及性分析。代表性组织包括:愈伤组织、胚根、胚芽、叶片、叶鞘、根、顶端分生组织(AM1/AM2)、休眠芽(DBuds)、茎端分生组织(SAM1/SAM2/SAM3)、穗颈节(PNN)、茎、幼穗(Panicle1–Panicle4)、外稃、内稃、雌蕊、雄蕊及种皮(Seed1–Seed3)。实验在 3 个代表性品种中开展:日本晴(NIP,粳稻亚种)、明恢 63(MH63,籼稻 II 型)和珍汕 97(ZS97,籼稻 I 型),每样本均设至少 2 次生物学重复(图 1a 与补充数据 1)。
总计获得 145 个高质量全基因组染色质可及性数据集,平均测序深度约 30.7 M reads。我们遵循 ENCODE 标准 13,14 搭建分析流程(见方法)。与 ChIP-Hub 数据库 14 中已发表的植物 ATAC-seq 数据相比,本研究数据信噪比显著提高(补充图 1)。利用三个品种各自参考基因组 15,16 进行分析,平均每实验鉴定 40,676 个(28,991–49,737 个)可重复 OCR(不可重复发现率 IDR 17 < 0.05)(图 1b)。
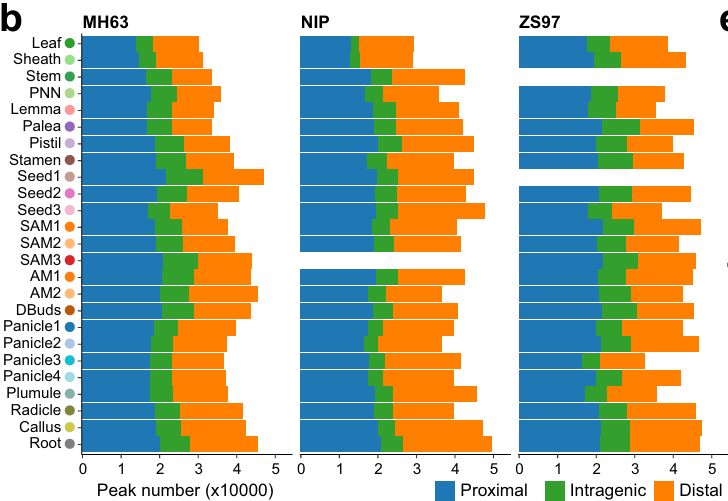
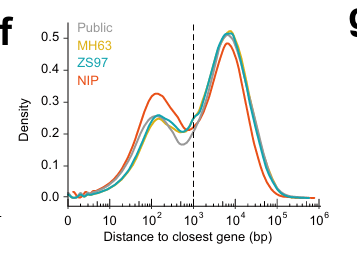
正如预期,所有实验检出的 OCR 主要集中于转录起始位点(TSS)近端上游区域或远端基因间区(图 1b, f;补充图 2;补充数据 2),分别对应启动子或增强子特征 18。值得注意的是,基因内部 OCR 占比较低(约 15.7%),且大多来源于内含子区域(补充图 2b)。这些结果表明,水稻基因组中的 OCR 绝大部分源自非编码序列。
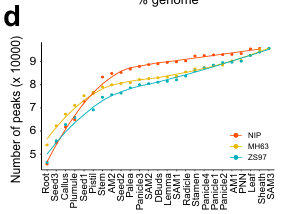
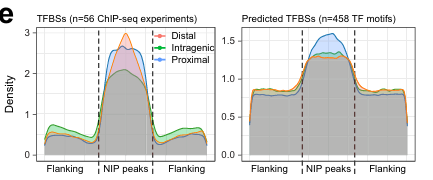
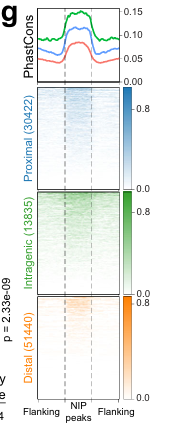
我们估计,大约 15% 的水稻基因组可被注释为开放染色质区域(OCR),且这一比例在三个品种中高度一致(图 1c);饱和度分析显示,该估计已趋于饱和(图 1d)。OCR 富含多种转录因子(TF)结合位点,并负责调控其靶基因的表达。我们收集了 56 种 TF 的公开 ChIP-seq 数据(补充数据 3),并结合 ChIP-Hub 数据库 14 中预测的 458 种水稻 TF DNA 基序,结果显示 OCR 显著富集 TF 结合位点(图 1e)。此外,与侧翼基因组区域相比,OCR 表现出更高的进化约束(图 1g),支持了先前关于保守非编码序列(CNSs)可预测植物 OCR 的发现。
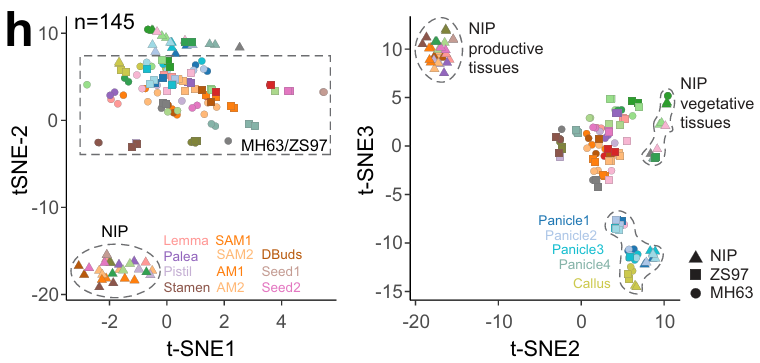
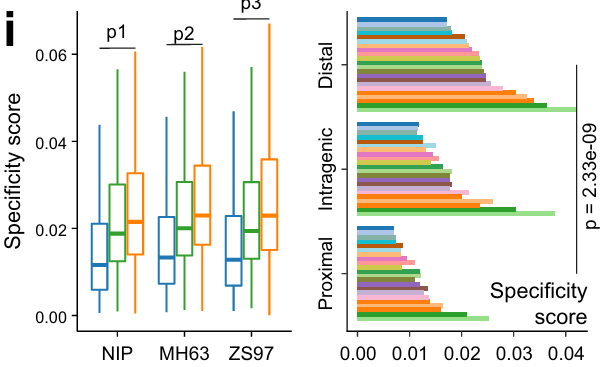
随后,我们从同一参考基因组(日本晴)合并全部 OCR(n = 117,176),对所有数据集进行量化,并利用 t-SNE 降维可视化其整体模式。结果中,t-SNE 的第 1、2 维主要反映籼稻(MH63 和 ZS97)与粳稻(NIP)亚种间的差异,而第 2、3 维则清晰区分不同组织类型(图 1h)。例如,日本晴的营养组织与生殖组织在染色质可及性上各自聚类;而幼穗与愈伤组织无论品种来源如何,均呈现相似的模式。我们进一步基于 Jensen-Shannon 散度(JSD)计算每个 OCR 的组织特异性,发现远端 OCR 的特异性得分显著高于近端 OCR(图 1i 及补充图 3a,b),与先前报道一致。
综上,本研究构建的水稻全景式开放染色质图谱,为作物功能基因组学研究提供了宝贵资源。
将开放染色质区域(OCR)与靶基因关联
为解析这些 OCR 可能调控的基因,我们为每个品种的全部受检组织同步测定了相匹配的 RNA-seq 数据(补充图 3c、补充数据 4)。我们采用现有策略 23,以所有样本中 OCR 可及性与基因表达的相关性为基础,预测 OCR–基因调控链接(图 2a;见方法)。基因可借助多个 OCR(包括启动子与增强子)通过染色质互作实现调控,而这些互作通常发生在拓扑关联结构域(TAD)内部。根据水稻 Hi-C 数据 24,25,TAD 大小估计为 35–45 kb,因此我们将搜索范围限定在转录起始位点(TSS)上下游各 20 kb(共 40 kb)以内,以预测 OCR 的潜在靶基因。
以绝对皮尔逊相关系数 |R| ≥ 0.4 且 P < 0.05 为阈值,我们共获得 59,075 条唯一的 OCR–基因链接,涉及 38,437 个 OCR(占全部 OCR 的 32.8%)与 18,781 个基因(占注释基因的 48.1%;补充图 4a,b 与补充数据 5)。正如预期,近端 OCR 的 OCR–基因链接频率显著更高,其基因表达与染色质可及性的相关性也随之增强(补充图 4c–f)。