ChatNT-用于DNA、RNA和蛋白质任务的多模态对话代理-文献精读143
A multimodal conversational agent for DNA, RNA and protein tasks
用于DNA、RNA和蛋白质任务的多模态对话代理
https://huggingface.co/InstaDeepAI/ChatNT
摘要
语言模型正在蓬勃发展,推动着对话代理的应用,帮助和赋能人类解决各种任务。最近,这些模型已被扩展以支持更多的模态,包括视觉、音频和视频,展示了在多个领域(包括医疗保健)中的卓越能力。然而,在生物学领域,对话代理仍然受到限制,因为它们尚未完全理解生物序列。与此同时,通过对序列数据进行自监督,已经构建了高性能的生物序列基础模型,但这些模型需要针对每个具体应用进行微调,导致任务之间的泛化能力有限。此外,这些模型并非对话型,这限制了它们对没有编码能力的用户的实用性。本文提出通过引入ChatNT来弥合生物学基础模型与对话代理之间的鸿沟,ChatNT是一种具有先进生物序列理解能力的多模态对话代理。ChatNT在Nucleotide Transformer基准测试中实现了新的最先进的结果,能够一次性解决所有任务(使用英语),并能对未见过的问题进行泛化。此外,我们还策划了一套更具生物学相关性的任务,涵盖DNA、RNA和蛋白质,跨越多个物种、组织和生物过程。ChatNT在这些任务上达到了与最先进的专门方法相当的性能。我们还提出了一种基于困惑度的技术,帮助校准模型预测的置信度。通过应用通过英语解码器和DNA编码器的归因方法,我们展示了ChatNT的回答基于生物学上连贯的特征,如检测启动子TATA基序或剪接位点二核苷酸。我们的基因组指令调优框架可以扩展到更多任务和数据模态(例如,结构和影像),使其成为生物学领域的广泛适用工具。ChatNT为构建能够理解生物学的通用智能代理提供了潜在方向,同时对没有编码背景的用户也具备可访问性。
正文
理解细胞、组织和生物体如何解读基因组中编码的信息,对于推进我们对生物学的理解至关重要。一个生物体的DNA序列包含了所有指定RNA和蛋白质的指令,同时也指示了这些分子应何时以及在何种细胞背景下产生。自从人类基因组被测序以来,主要的研究重点一直是识别每一个基因组元素,表征它们的功能,并评估遗传变异对不同基因调控和细胞过程的影响。考虑到生物序列和过程的复杂性以及基因组学数据的日益增加,已经开发出几种机器学习和深度学习方法,以极高的准确性预测不同的分子表型。这些任务包括预测蛋白质与DNA和RNA的结合、DNA甲基化、染色质特征、调控元件、剪接、基因表达、mRNA特性(如稳定性和多腺苷酸化)以及蛋白质特性(如熔点)等。
尽管监督学习的深度学习模型已经显著提高了这些任务的预测能力,但由于标签数据的稀缺性(标签过程耗时且昂贵),它们的性能仍然常常受到限制。然而,由于现代测序技术的通量增加和成本降低,大量未标记的基因组数据正在快速增加,这为自监督深度学习方法提供了一个重要机会,利用这些未标记的数据进行训练。通过采用如掩码预测或下一个标记预测等学习技术,深度学习模型可以在预训练阶段构建强大的基因组基础表示,聚合核苷酸和较大序列模式之间的关联,形成富有表现力的高维向量,捕捉已知的基因组元素和蛋白质结合位点。这些模型可以在微调阶段利用这些丰富的表示,学习得更快,并在监督任务中取得更好的性能,即在有标签的数据上进行训练,即使数据稀缺。最近,已有多种此类基础模型以这种方式构建,并展示了它们可以在成百上千个物种的基因组上进行预训练,然后微调以解决大量的分子表型预测任务。
尽管如此,当前DNA基础模型的性能和应用领域仍然有限。在当前的范式中,基础模型需要为每个特定任务单独进行微调,以生成准确的表示和预测,因此更适合作为特定任务的专家。这不仅导致随着任务数量的增加,模型的数量也急剧增加,而且还阻碍了监督任务之间的迁移能力以及在零样本环境下(即无需对某些示例进行进一步微调)解决新任务的能力。因此,亟需重新思考基因组学人工智能(AI)系统的开发,目标是建立通用的统一模型,捕捉所有不同生物序列和功能之间的复杂关系。其他领域,如自然语言处理(NLP)和计算机视觉,已经证明并行训练多个任务可以实现任务之间的知识迁移,并提高准确性和泛化能力。在这些领域中,英语语言被证明发挥着更广泛的作用:作为一种通用接口,用于表示各种任务和指令,并帮助指导端到端多任务模型的训练。将这种方法转移到生物学数据上,是开发可以同时解决所有基因组学任务并提高准确性的通用模型的一种有前景的方法。
构建通用基因组学AI系统的另一个重要方面是其对不同类型用户的可访问性。大多数生物学家不知道如何使用当前的基因组学模型,更不用说如何为特定任务编程一个模型了。此类模型不是对话型的,因此对于没有编码能力的用户来说,其实用性有限。在这里,语言作为一种通用接口,能够发挥重要作用,作为一个通用AI助手,通过能够明确表示为英语语言的任务指令来解决基因组学任务。例如,最近ChatGPT和GPT-4的成功展示了大型语言模型(LLM)在跟随人类指令方面的强大能力,以及这种工具如何通过其易用性转变多个行业。我们设想,一旦我们拥有像ChatGPT一样精通生物学任务的代理程序,基因组学和生物学领域也会发生同样的范式转变。
为此,我们在本文中提出了一种构建基因组学基础模型的方法。与NLP领域的研究成果类似,并受到近期视觉和语言多模态模型的启发,我们建议将所有监督基因组学预测任务表述为文本到文本的任务,并构建一个多模态DNA和语言代理,命名为Chat Nucleotide Transformer(或ChatNT)。ChatNT可以接收一个或多个DNA序列,并用英语提示来解决所有这些任务。这种表述方法使我们能够使用相同的词汇(即英语和DNA词汇的拼接)来表达所有任务,并通过最小化统一目标来学习解决这些任务,类似于GPT类模型,从而实现无缝的新任务集成和泛化。用英语表述任务也是向模型提供附加元数据(如物种、染色体或细胞类型)的简单方式,而这些信息在大多数现有的DNA基础模型中是缺失的。
ChatNT被构建为一个通用的基因组学AI系统——一个可以解读多种生物序列并处理数十个任务的统一模型,在对话代理设置中运行。我们创建了基因组学指令任务数据集,包含了多种分类和回归任务的英语问题和指令集。我们首先展示了ChatNT在Nucleotide Transformer基准测试中取得了新的最先进成果。接下来,我们在更多与生物学相关的任务中评估了ChatNT,这些任务涵盖了DNA、RNA和蛋白质过程。ChatNT在所有任务中都达到了最先进的性能,与多个专门模型的性能相匹配,例如用于RNA多腺苷酸化的APARENT2和用于蛋白质相关任务的ESM2,同时能够一次性解决大量任务并以英语进行表达。最后,其英语对话能力使其比其他模型更易于使用,扩大了其对没有机器学习或计算机科学背景的科学家的可访问性。我们的基因组学指令调优框架可以轻松扩展到新的任务或生物数据模态(例如,测序实验和影像),无需每次从头开始预训练,使其成为一个广泛适用于生物学的工具。
结果
将DNA基础模型转化为对话代理——ChatNT
ChatNT是一个基因组学指令调优框架,它将指令调优代理扩展到生物学和生物序列的多模态空间。我们的框架设计为模块化并且可端到端训练。它结合了以下三部分:(1) DNA编码模型,预训练于基因组测序数据,提供DNA序列表示;(2) 英语解码器,通常是预训练的GPT风格的大型语言模型(LLM),用于理解用户指令并生成响应;(3) 一个投影层,将DNA编码器提取的表示投影到输入的英语单词嵌入空间中,使得这两者都能被英语解码器使用(见图1c和方法部分)。与大多数多模态研究(例如,参考文献26)通常会冻结编码器,仅训练投影层,有时还会训练解码器不同,在本研究中,我们决定除了投影层外,还要对编码器进行反向传播,以允许在DNA模型层级进行监督知识传播。由于英语解码器保持冻结,ChatNT能够受益于其完整的初始对话能力,确保这些能力在训练过程中不会退化。在本研究中,我们使用Nucleotide Transformer v2(一个在850个物种基因组上预训练的5亿参数模型)作为DNA编码器,以及Vicuna-7b(一个经过指令微调的7亿参数LLaMA模型)作为英语解码器,来构建对话代理ChatNT。保持这种模块化架构使得将来可以在不改变模型架构的情况下,使用不断改进的编码器和解码器。
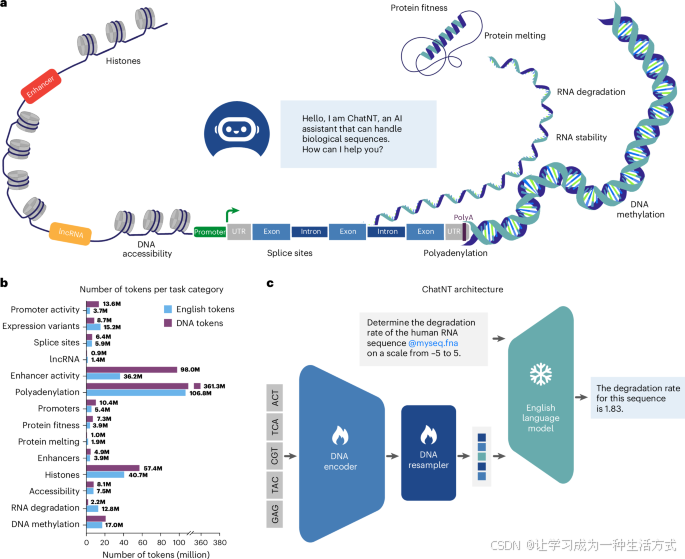
将DNA基础模型转化为对话代理——ChatNT
a, 训练过程中所包括的不同类别下游任务的示意图。UTR,未翻译区。
b, 我们的基因组学指令数据集中,每个任务可用的英语和DNA标记的统计数据。英语问答指令使用LLaMA分词器进行标记化,而DNA序列则使用Nucleotide Transformer分词器进行标记化。
c, ChatNT构建多模态和多任务基因组学AI系统的方法。ChatNT对话代理可以通过英语提示来解决给定问题和核苷酸序列的各种任务。在这个例子中,用户输入一个DNA序列(fasta文件),并要求代理评估给定RNA序列的降解率。问题标记与投影后的DNA表示结合后,传递到英语语言模型解码器中。预训练的解码器通过下一个标记预测写出答案,在此情况下,预测输入序列的降解率。
为了训练和评估ChatNT,我们将基因组学任务数据集转换为指令数据集,通过用英语框定每个任务(参见补充图1,方法和“结果”部分)。我们为每个任务创建了训练和测试文件,包含各自的DNA序列,并结合了经过整理的英语问题和答案。参见图1c,预测RNA降解水平的问答示例:‘用户:在-5到5的范围内,确定人类RNA序列@myseq.fna的降解率。ChatNT:该序列的降解率为1.83’,其中候选DNA序列的投影嵌入插入在@myseq.fna位置。我们保留了每个任务原始数据的相同训练-测试划分,并使用不同的问题进行训练和测试,以评估模型的英语泛化能力。这不仅使得可以评估代理在DNA序列间的泛化能力,还可以评估其对所使用英语语言的鲁棒性。我们还提供了一种灵活的方法,通过使用位置标签(@myseq.fna)交错英语和DNA序列,使用户能够在同一问题中引用多个序列。
ChatNT被训练为同时解决所有任务,在每个批次中对任务进行统一抽样。ChatNT通过自然语言提示实现多任务处理,用户提出的问题将指导代理进行感兴趣的任务。给定文本提示和一个或多个DNA序列作为输入,ChatNT被训练以最小化所有任务的统一目标,该目标表现为ChatNT预测与目标答案标记之间的交叉熵损失,类似于其他指令微调工作的做法。这个单一的目标使得在不同任务间无缝学习成为可能,而不会引入来自不同目标和损失函数(例如,分类的交叉熵与回归的均方误差(MSE))的冲突梯度或尺度问题。此外,它还允许我们在未来扩展模型以处理额外的任务,而无需改变模型架构或从头开始训练。总之,ChatNT提供了一个通用的基因组学AI系统,以对话方式解决多个任务,从而为基因组学模型提供了不同的范式。
除了无缝整合多种类型的标记和实验数据到单一的通用基础模型中,ChatNT还被设计为对话式的,使用户能够轻松与其交互,并在不需要编程背景的情况下使用它(参见补充图1中的示例)。我们依赖一个冻结的英语语言模型Vicuna-7b,它是从LLaMA模型微调得到的。ChatNT保持了语言模型的所有固有对话能力。有趣的是,我们观察到,由于用于构建LLaMA的训练数据集已经包含了大量生命科学论文,我们的代理还能够回答关于基因组学的多个问题,包括定义调控元素如启动子和增强子,零-shot,即无需任何额外的训练数据。此外,ChatNT还能够回答许多与生物学无关的问题,并解决诸如摘要或编写简单编程代码等任务。由于我们的方法是通用的,并且建立在任何预训练的英语语言模型之上,ChatNT的能力可以随着新的、更强大的开源语言模型的出现而自然改进。虽然对话能力是ChatNT的一个重要方面,但已经由相应的语言模型提供,我们在这项工作中集中展示了ChatNT能够以英语高精度解决一系列高级基因组学任务。
ChatNT在基因组学基准测试中取得了改进的性能
为了开发ChatNT并优化其架构,我们创建了Nucleotide Transformer基准测试的指令版本(方法和补充表1)。这个基因组数据集适用于在模型实验期间的快速迭代,因为它包含了一组多样的小型数据集,并且已经在多项DNA基础模型研究中进行了广泛评估。我们训练ChatNT一次性解决所有18个任务,并使用测试集中的DNA序列和问题评估其性能。
我们首先使用这个基准测试,系统地比较了ChatNT与两种不同投影架构的性能。在之前的多模态模型中,聚合编码器信息的经典方式是使用可训练的投影,将编码器嵌入转换为语言嵌入标记,这些标记的维度与语言模型中的单词嵌入空间相同。在ChatNT中,我们使用了来自Flamingo的Perceiver重采样器,基于门控交叉注意力作为投影层(参见补充图2a)。使用这种投影层,并在所有18个任务上微调DNA编码器和投影,ChatNT在这个基准测试中取得了新的最先进准确率,平均Matthew相关系数(MCC)为0.71,比之前的最先进模型Nucleotide Transformer v2(500M)高出2个点(图2a和补充图2d和3)。
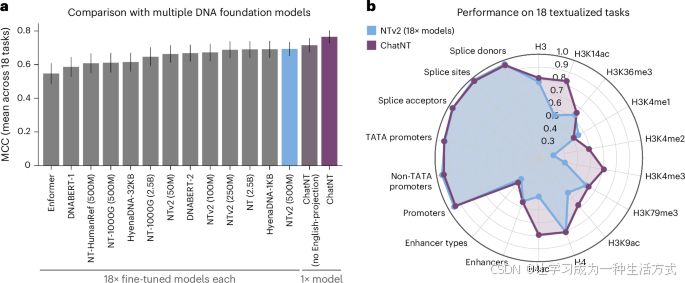
a, ChatNT、没有英语感知投影的ChatNT和13种不同基因组学基础模型在Nucleotide Transformer基准测试中的18个任务上的平均表现。条形图表示平均MCC值±标准误(n=18)。
b, 一个雷达图,展示了ChatNT在18个任务中的表现,与专门微调于每个任务的Nucleotide Transformer v2模型的比较。
然而,类似于所有其他投影层,当前Perceiver重采样器的实现为编码器标记生成相同的固定嵌入集,无论所提问题如何,因此它需要在这组嵌入中捕捉每个下游任务的所有相关信息。我们假设,鉴于潜在序列的多样性(包括不同的长度、物种和生物学属性),这个特性可能在扩展模型以应对多个下游任务时,产生基因组学中的信息瓶颈。因此,我们开发了一种英语感知的Perceiver投影,它从输入序列中提取与用户提问相关的表示,这使得能够利用输入DNA序列中对特定问题相关的上下文信息(方法和补充图2b)。我们观察到,在将DNA嵌入投影到英语解码器空间时,通过考虑问题显著提高了性能(平均MCC为0.77,较0.71有所提升;图2a和补充图2c、d)。这可以通过DNA序列中的非常具体的上下文和任务特定信息来解释,必须保留这些信息才能解决各种基因组学任务。由于解码器保持冻结,投影层不仅需要将序列嵌入带入英语解码器的嵌入空间,还需要执行操作,从嵌入中提取相关信息以回答问题。我们的结果表明,使投影感知问题有助于这两个方面,从而实现更好的性能和任务间的迁移。
总之,具有英语感知投影的ChatNT(以下简称ChatNT)在此基准测试中达到了新的最先进准确度(平均MCC为0.77),并且一次性解决了所有18个任务(图2a)。ChatNT比之前的最先进Nucleotide Transformer v2(500M)模型提高了8个点(作为ChatNT中的DNA编码器,平均MCC为0.77对比0.69;图2a、b)。除了在DNA序列和任务之间进行泛化,ChatNT还能够在问题之间进行泛化,正如基于语言提示变化的预测准确度的低变异性所展示的(补充图4)。我们的结果表明,通过自然语言制定的单一统一目标促进了多个下游任务之间的迁移学习,并帮助提供了改进的性能。
精心策划的生物学相关任务指令数据集
尽管Nucleotide Transformer基准测试非常适合模型实验和调试系统,但它遗漏了许多在基因组学中具有重要生物学意义的任务,特别是涉及更复杂生物学过程以及更现代实验技术和涉及定量预测的任务。因此,我们精心策划了第二个基因组学指令数据集,包含27个以英语表述的基因组学任务,这些任务来源于不同研究,涵盖了多个调控过程(方法和补充表2)。这些任务包括与DNA(21个任务)、RNA(3个任务)和蛋白质序列(3个任务)相关的任务,涵盖了多个物种,并框定为二元/多标签分类任务和回归任务。最终的指令数据集包含了总计6.05亿个DNA标记(即36亿个碱基对)和2.73亿个英语标记(包括每个任务平均1000个问答对)(图1b)。
该集合包括来自Nucleotide Transformer和BEND基准测试的非冗余任务子集,并补充了来自植物AgroNT基准测试和人类ChromTransfer的相关任务。这些基准测试在文献中得到了广泛使用,来自不同的研究小组,代表了多种DNA过程和物种。这些选定的任务包括涉及组蛋白和染色质特征、启动子和增强子调控元素、剪接位点等生物过程的二元和多标签分类任务。
我们进一步添加了与启动子活性、增强子活性、RNA多腺苷酸化和降解,以及多种蛋白质特性相关的最先进且具有挑战性的回归任务。这些数据集是各自领域的参考数据集,涉及生物学DNA、RNA和蛋白质序列的复杂特性。所有RNA和蛋白质任务都是从相应的DNA和编码序列(CDS)预测的,而不是直接从RNA和蛋白质序列预测的。对于RNA序列,获取匹配的DNA序列很简单,但对于蛋白质序列,由于密码子使用的复杂性,这更加具有挑战性。因此,我们使用了Boshar等人策划的CDS注释来处理蛋白质任务。
参见图3和补充图1,了解我们数据集中不同类型基因组学任务的问答示例(还可参见补充图5–7)。例如,启动子分类任务的训练示例如下:“用户:在人类细胞中,序列@myseq.fna中是否存在启动子,能否判断其强弱?ChatNT:是的,在您提供的DNA序列中存在一个弱启动子。”其中候选DNA序列的投影嵌入插入在@myseq.fna位置。回归任务也以英语表述,代理需要写出对应所请求数量的数字:例如,“用户:在-5到5的范围内,确定小鼠RNA序列@myseq.fna的降解率。ChatNT:该序列的降解率为2.4。”(定量刻度的详细信息见方法部分)。损失计算与交叉熵损失相同,将标量值视为数字标记。这种方法在我们的设置中表现良好,并且产生了与传统MSE损失相当的结果,因为ChatNT是自回归的,可以捕捉数字的顺序结构。较早的数字错误(例如,数量级顺序)会导致更高的损失,促使模型首先集中于预测最重要的数字,然后再精细调整较不重要的数字,从而有效地引入了数字的层次分解。为了评估性能,我们提取每个答案中的数字,并测试它们与真实值的相关性。
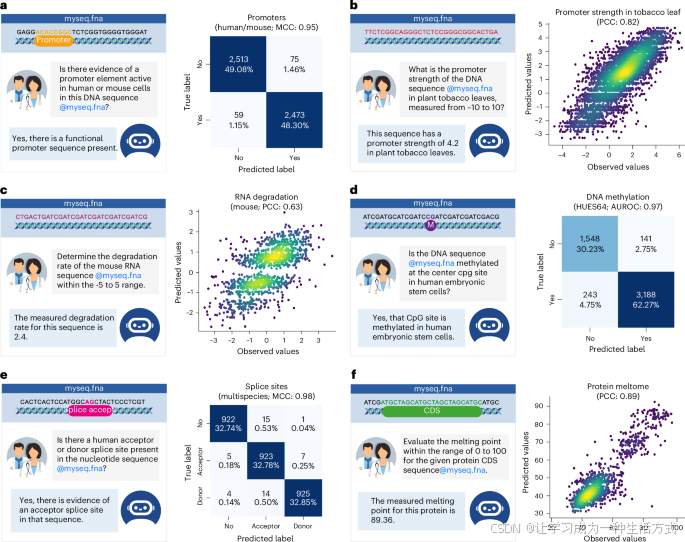
a, d, e,左侧:启动子(a)、DNA甲基化(d)和剪接位点任务(e)的对话示例。右侧:显示混淆矩阵的热图,比较ChatNT预测的标签与观察到的标签。报告了性能指标。
b, c, f,左侧:烟草叶片中的启动子强度(b)、RNA降解(c)和蛋白质熔解组任务(f)的对话示例。右侧:散点图,比较ChatNT的预测值和观察值。报告了皮尔逊相关系数(PCC)。
总之,这组精心策划的任务为我们模型在不同生物序列领域中的能力和实用性提供了一个总体视角。我们将ChatNT训练为一个通用代理,能够一次性解决所有27个基因组学任务,并以英语进行表达,并将其性能与每个任务的最先进专用模型进行了比较(方法部分)。
在不同基因组学过程和物种任务上的评估
我们首先评估了ChatNT在与不同DNA过程相关的21个任务上的表现,这些任务涉及酵母、植物、果蝇、小鼠和人类。ChatNT在与不同专用模型进行直接微调的这些任务上表现出色,并且对语言提示的变化具有较强的鲁棒性(图3a、b、d、e和图4a、c)。特别地,我们在检测人类增强子类型方面取得了改进的表现。然而,我们也观察到,与专门在该任务上进行微调的最先进的AgroNT模型相比,植物物种的增强子性能显著下降。由于AgroNT是基于48种不同植物物种的基因组进行预训练的,因此改善ChatNT中使用的编码器可能会提高这种类型任务的性能。
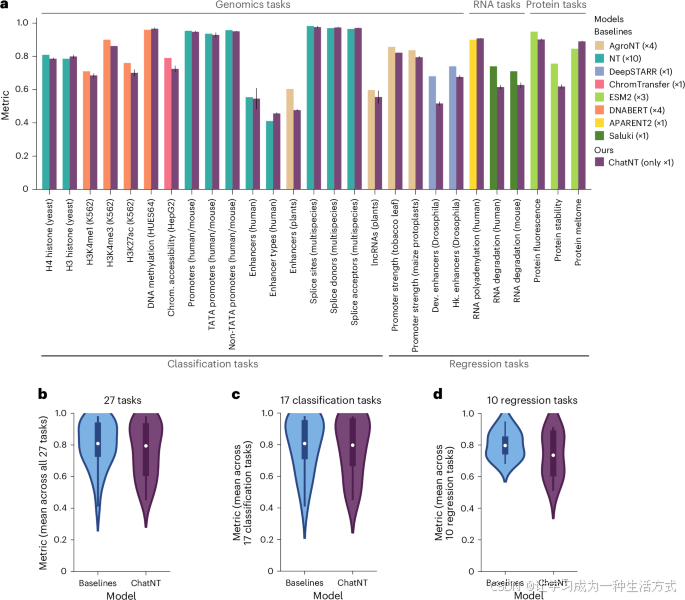
a,条形图显示了ChatNT与每个任务对应基准的性能比较。每个任务使用的度量与相应基准研究中使用的度量相同(补充表2)。数据以均值±标准差(n > 10每个任务)表示。Chrom.,染色质;Dev.,发育;Hk.,管家基因。
b–d,ChatNT与基准模型在所有任务(n = 27)(b)、分类任务(n = 17)(c)和回归任务(n = 10)(d)上的比较。度量与a中相同。箱线图标出中位数、上四分位数和下四分位数,以及1.5倍四分位范围(须状线);离群值单独显示。
由于ChatNT以英语解决任务,它能够无缝处理二元和多标签分类任务。通过提取ChatNT在答案中预测的术语,我们可以量化其预测性能。正如我们在图3中的一些示例所展示的,ChatNT准确识别了包含人类或小鼠启动子的输入序列(图3a),包含在人类胚胎干细胞(HUES64细胞系;图3d)中甲基化的CpG位点的序列,以及包含剪接受体和供体位点的序列(图3e)。
ChatNT还能够通过写出预测得分的数字来解决定量任务。我们观察到,在预测植物启动子活性方面表现具有竞争力,特别是在烟草叶片(图3b)和玉米原生质体中,但在果蝇增强子活性方面与最先进的DeepSTARR模型相比,性能显著降低(图4a)。重要的是,预测数字的分布与原始得分之间有很好的相关性(图3b)。这一有效处理回归任务的能力在生物学中至关重要,尤其是在考虑到语言模型在数值处理方面的已知局限性和不可靠性时。这一点特别重要,因为回归任务相较于分类任务,在复杂性上有所不同,分类任务在训练集中的比例更高。我们认为,这可以通过在训练过程中改善分类任务和回归任务之间的平衡来解决,可以通过加权损失或任务抽样频率课程来实现。
ChatNT解决转录组学和蛋白质组学任务
ChatNT采用灵活的架构,允许其处理任何可以用我们的DNA编码器——Nucleotide Transformer处理的生物序列。为了展示其泛化能力,我们在新的基因组学指令数据集中包含了三个RNA和三个蛋白质回归任务(补充图6和7)。这些任务包括预测RNA多腺苷酸化和降解速率,以及不同蛋白质特性。用于模型训练的对话示例如下:“用户:在人类HEK293细胞中,RNA序列@myseq.fna的近端位点的多腺苷酸化比率是多少,范围从0到1?ChatNT:该序列的近端位点多腺苷酸化比率为0.69。”和“用户:指定给定编码序列(CDS)@myseq.fna的蛋白质熔点,范围在0到100之间。ChatNT:该蛋白质的熔点为80.81。”。ChatNT的性能与最先进的专用模型进行了比较,包括用于多腺苷酸化的APARENT2模型、用于RNA降解的Saluki模型和用于蛋白质任务的ESM2模型(补充表2)。
总体而言,我们在六个RNA和蛋白质任务的测试集上观察到ChatNT表现良好,皮尔逊相关系数(PCC)在0.62到0.91之间(图3c、f和图4a)。ChatNT在预测近端多腺苷酸化位点比率(PCC为0.91,对比0.90)和蛋白质熔点(PCC为0.89,对比0.85)方面超越了专用模型。对于人类和小鼠的RNA降解任务,ChatNT的PCC为0.62和0.63,比专用的Saluki模型低了10个点(PCC为0.74和0.71)。ChatNT在与最先进的蛋白质语言模型ESM2的两个蛋白质任务(与蛋白质荧光和稳定性相关)上的表现也具有竞争力。尽管ChatNT目前还无法超越每个专用模型在RNA和蛋白质任务上的表现,但我们展示了它已经能够处理这些任务,并且利用DNA基础模型Nucleotide Transformer作为DNA编码器,取得了高性能。ChatNT的灵活架构允许用户插入不同的编码器,如专门用于RNA和蛋白质领域的语言模型,这应能缩小RNA和蛋白质组学领域与专用深度学习模型之间的差距,并提高ChatNT作为统一生物学模型的能力和泛化能力。
评估ChatNT答案的可信度
ChatNT旨在帮助和增强科学家和研究人员的日常研究。因此,它的性能和可靠性至关重要。然而,与返回概率或定量得分的标准机器学习模型不同,ChatNT直接回答问题,无法让用户感知其置信度,从而减少其在敏感应用中的实际价值。这是一个重要挑战,并且是当前所有对话型代理共同面临的问题。为了解决这个问题,我们研究了一种评估我们代理在二元分类任务中置信度的方法。我们不是直接为给定序列生成二元分类问题的答案,而是计算该问题的模型困惑度,基于正面和负面答案的示例。我们确保这些选定的答案不包含在模型的训练数据集中。这些困惑度值对于正面和负面答案的计算结果将用于推导每个类别的logits和概率。该方法使我们能够像标准分类器一样从ChatNT推导每个问题示例的概率,我们将其称为基于困惑度的分类器(图5a)。目前,这个过程被作为事后分析进行研究,尚未集成到ChatNT工具中,但一旦完全开发出来,可以在未来的版本中加入,提供这些信息给用户。
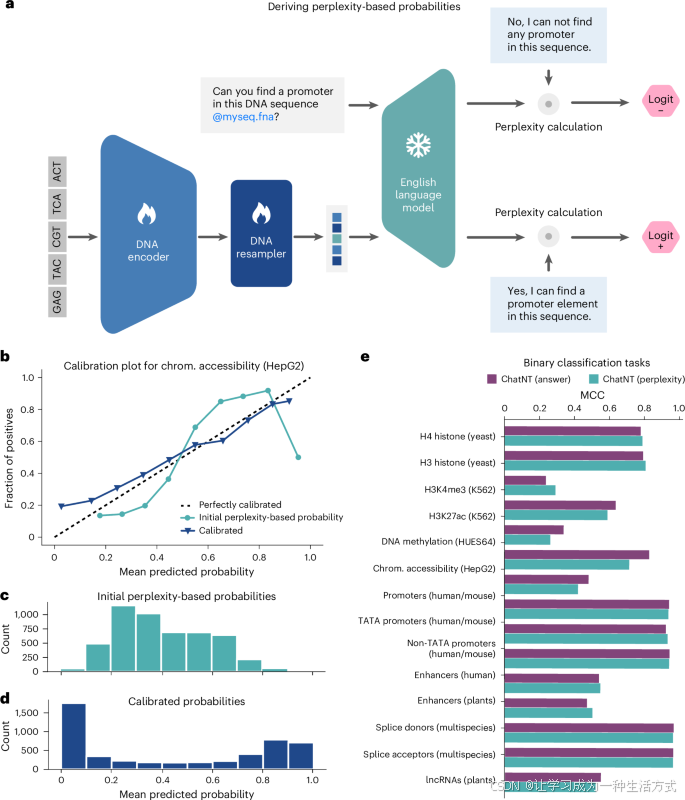
a,描述基于困惑度分类器的卡通图,基于ChatNT的回答。
b,人类染色质(chrom.)可及性任务的校准图(HepG2细胞系)。散点图比较了原始(绿色)和校准(紫色)基于困惑度的分类器的预测概率与阳性样本的比例,在十个区间中的表现。
c,原始基于困惑度的分类器在十个区间内的预测概率直方图。
d,校准后的基于困惑度的分类器在十个区间内的预测概率直方图。
e,ChatNT答案(是与否)与其派生的基于困惑度的概率在所有二元分类任务中的性能比较(MCC)。
计算概率使我们能够评估模型的校准性,即预测概率、其置信度与预测准确性之间的相关性。当一个模型在给定置信度为p时,预测的类别总是100%正确时,我们称该模型为良好校准的。我们计算了ChatNT在所有二元分类任务中的基于困惑度的概率。在图5b–d中,我们展示了基于染色质可及性任务的预测结果的校准图示例。我们观察到,我们的模型在低置信度和高置信度区域校准良好,但在中等置信度区域表现较差。例如,预测概率为0.9的示例正确预测的概率为90%,而预测概率为0.5的示例仅有25%的时间正确预测。为改善这一点,我们展示了通过在训练集上拟合Platt模型来校准我们的模型,从而提高模型在所有预测范围内的置信度(图5b–d)。此校准步骤适用于所有二元分类任务。总体而言,使用这些基于困惑度的预测,ChatNT在各个任务上的性能保持一致(图5e),但校准性得到了提高。因此,我们的方法不仅可以准确衡量语言模型的预测性能,还能有效评估其不确定性水平。这项技术,尽管是通用的,应该对其他语言模型领域也有益。
模型解释揭示了学习到的DNA序列特征
语言模型在生物序列中的一个关键应用是揭示DNA、RNA和蛋白质序列的潜在编码或语法。为了评估ChatNT的预测是否依赖于生物学相关的序列特征,并且是否可以用于进一步的发现,我们使用了模型解释工具分析来自我们数据集中不同任务的数百个序列。虽然这些技术已广泛应用于序列模型,特别是在DNA的情况下,但在输出以自然语言表达的多模态模型中,如ChatNT,这种做法尚未被应用。为了实现这一点,我们量化了输入DNA序列中每个DNA标记(即6-mer)对ChatNT预测的英语标记(回答)的贡献(图6a)。更具体地,我们计算了预测标记相对于输入DNA标记的梯度,通过英语解码器、Perceiver投影和DNA编码器进行反向传播。我们将这种方法应用于三个不同基因组学任务的序列,这些任务在文献中已知具有明确的预测序列特征:通常与GT二核苷酸重叠的剪接供体位点、与AG二核苷酸重叠的剪接受体位点以及依赖于TATA-box基序的启动子。
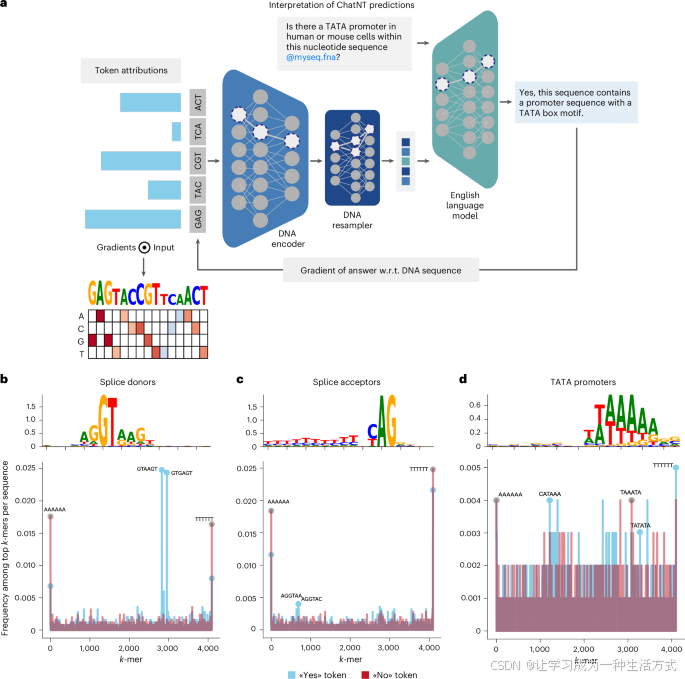
a,描述ChatNT预测的解释卡通图,针对输入序列。对于给定的输入问题和DNA序列,我们计算每个预测的英语标记对每个输入DNA标记的梯度。w.r.t.,相对于。
b–d,从ChatNT中提取的序列特征,任务为剪接供体(b)、剪接受体(c)和TATA启动子(d)。展示了合并的序列基序,y轴为比特(上),每个任务的序列k-mer谱图(下)。
对于每个任务,我们计算了输入DNA标记相对于ChatNT所回答的“是”或“否”标记的贡献。我们对每个任务的每个标签的测试集序列子集进行了此分析,识别出每个输入序列中贡献最大的DNA标记。显示重要的6-mer在所有序列中的频率,揭示了正标签序列在已知的序列特征上有所富集,例如剪接供体和受体二核苷酸(图6b–d)。实际上,通过结合所有正序列中得分最高的标记并计算每个位置的核苷酸频率,恢复了每个任务的主要序列模式,匹配了已知的剪接供体GT二核苷酸、剪接受体AG二核苷酸和启动子TATA基序。总的来说,这些结果表明,ChatNT的答案基于生物学上连贯的特征,并且该模型可以用于解释DNA的基础语言。值得强调的是,ChatNT通过单一的统一模型完成这一任务。通常,这种分析依赖于为每个任务量身定制的专用或微调模型。我们的结果揭示了在不同任务中提取的一致且有意义的特征,凸显了ChatNT学习DNA通用语法的能力,并将其与自然语言连接。
讨论
我们提出了ChatNT,一个多模态对话代理,可以处理DNA、RNA和蛋白质序列,并解决多个生物学相关的下游任务。我们构建并策划了基因组学指令任务数据集,包括二元和多标签分类及回归任务,涵盖了不同物种和基因组学过程。还包括与转录组学和蛋白质组学过程相关的任务,展示了这种方法在不同领域的多功能性和通用性。ChatNT在Nucleotide Transformer基准测试中达到了新的最先进水平,并在我们的27个任务数据集上展示了与专用模型相媲美的性能。重要的是,与需要为每个任务创建专用模型的常规方法不同,ChatNT在一个统一的模型中解决了所有任务,并提供了一个简单自然的聊天界面,供用户使用该模型。我们还引入了一种技术来探测语言模型在二元分类任务中的置信度,并在需要时进行校准。通过模型解释分析,我们还展示了ChatNT在回答不同基因组学任务时,学会了关联生物学相关的序列特征。总之,ChatNT证明了自然语言大型语言模型(LLMs)可以扩展到处理生物序列模态,不仅显示出对话能力,还能准确回答多个生物学相关的问题。
为了从DNA序列中提取解决所有任务所需的复杂信息,我们引入了基于Perceiver重采样器的架构,重新采样并将DNA嵌入投影到自然语言嵌入空间中。我们识别出一个信息瓶颈问题,这个问题源于任务、物种和生物过程的多样性,这些信息被编码在DNA序列中,并展示了如何通过根据所提问题对投影进行条件化来解决这一问题。这种条件化使投影模块能够从DNA嵌入中提取出适当的量,以解决当前任务,正如我们通过相较于未条件化投影模块的改进性能所展示的。
在这项工作中,我们决定专注于用户(如研究人员或科学家)希望检测分子表型或计算给定DNA序列的定量特性的情况。虽然我们相信这涵盖了已经相当多的实际应用案例,但将代理能力扩展到处理其他典型生物信息学管道将是很有趣的。这些管道可能包括调用工具来计算序列的统计数据,将序列与参考数据库进行比对以计算多序列比对,查询外部数据库获取关于序列的额外信息,或递归地调用ChatNT模型处理包含多个序列的FASTA文件并生成汇总的表格结果及其对应的分析。这得到了外部工具在大型语言模型中的成功支持,如Toolformer、LLaVA-Plus、geneGPT或GPT-4等。此类管道还可以从ChatNT处理多个序列的能力中受益,从而减少推理计算成本。通过将ChatNT当前的英语解码器替换为更大的模型和/或使用强化学习人类反馈微调的模型,如Llama2-chat 70B,可能有助于扩展模型在这些方向上的能力,并改善其整体实用性。
关于ChatNT可以处理的序列长度,代理具有其DNA编码器的固有限制。当前版本使用的是Nucleotide Transformer v2模型作为DNA编码器,该模型已经在最多12 kb长的序列上进行了训练。然而,存在多种技术可以用来扩展其上下文,多个研究表明,Nucleotide Transformer模型在处理高达50 kb的序列时表现有所提升。此外,可以在该框架中更换DNA编码器,例如使用Borzoi,它可以处理长达524 kb的序列,并使用任务所需的长上下文。未来,ChatNT可以通过使用新开发的DNA序列编码器来进一步改进。
ChatNT通过自然语言提示隐式处理物种和细胞类型信息,而不是作为结构化输入。当查询指定了物种或细胞类型时,模型直接从文本中提取并整合此上下文信息。这种方法利用了预训练语言模型根据语言上下文解释和条件化预测的能力。此能力通过我们的指令调优过程得到了促进,其中包括特定任务的示例,涵盖了不同的物种和细胞类型。将更多具有多样化提示的数据纳入训练应能使模型在广泛的生物学背景下进行泛化。一旦我们进一步扩大任务和数据的规模,未来该代理的一个额外应用场景将是零-shot预测基因组变异的功能影响。可以使用本研究中引入的位置标签来提示用户在同一个问题中引用多个序列。例如,用户可以使用两个不同的位置标签向ChatNT提供参考序列和突变序列,并让模型利用这两个序列的完整投影嵌入来进行突变影响的最终评估。一个例子是:“用户:我有一个突变,将序列@wildtype.fna更改为@mutation.fna。ChatNT:是的,这个基因变异是致病的。”这将为比较不同序列并通过DNA序列的学习表示评估基因变异的影响开辟新机会。
ChatNT的能力已通过使用预训练的DNA基础模型Nucleotide Transformer进行DNA序列处理得到展示。正如我们在实验中所示,处理DNA序列不仅可以解决基因组学中的任务,还可以解决转录组学和蛋白质组学中的任务,后者使用对应的CDS区域。然而,我们的方法可以轻松扩展,以集成来自其他组学模态的编码器,如RNA和蛋白质语言模型,以原生处理RNA和氨基酸序列。通过我们的位置标签系统,支持多个序列,可以简单地添加任意数量的编码器,并训练其相应的投影,以在同一问题中结合不同的组学和模态。我们设想这种方法可以进一步扩展我们模型的能力和性能,实现跨模态的优越迁移学习。
最后,我们注意到,目前ChatNT尚未建立机制来通知用户任务和细胞类型的覆盖范围(例如,训练数据中包含的内容)或当用户提出超出训练分布的问题时的处理方式。按照当前LLM研究的方向,这类机制可以通过向ChatNT的系统消息添加其训练的任务和数据集类型以及如何在提问超出该范围时处理来轻松实现。这些机制还应有助于防止滥用并确保这种工具的安全使用。
本工作作为一个概念验证,证明了可以构建多模态生物序列和英语对话代理,解决先进的生物学相关任务,并为构建未来理解生物序列和原理的高度能力代理奠定了基础。类似于自然语言处理(NLP)和多模态模型的发展,我们期望通过在两个主要方向上的发展,出现如零-shot性能等新能力:(1) 通过包括来自多样生物过程、组织、个体和物种的示例,扩大任务的数量;(2) 集成更多的数据模态,如RNA和蛋白质序列、影像数据和个人健康记录。当这些能力出现时,评估模型的安全性和鲁棒性将是至关重要的,例如通过红队测试。因此,ChatNT代表了向生物学和医学通用人工智能迈进的重要一步。
方法
ChatNT模型
架构
ChatNT是一个多模态代理,输入一个或多个DNA序列和一个英语提示,并返回一个英语单词的分布,用于自回归地生成一个英语答案。我们引入了一个DNA英语标记占位符<DNA>,它被添加到输入的英语提示中,供用户引用DNA序列。该架构也扩展为处理多个DNA序列。在这种情况下,每个DNA序列由DNA编码器独立处理,输入的英语提示预计包含与输入的DNA序列数量相等的DNA英语标记占位符。
ChatNT架构由三部分组成:一个预训练的DNA编码器,一个将DNA嵌入投影到英语单词嵌入空间的投影模型,和一个预训练的英语解码器。虽然我们的架构是通用的,理论上可以与任何DNA编码器和英语解码器搭配工作,但我们决定分别使用预训练的Nucleotide Transformer v2(一个在850个物种基因组上预训练的5亿参数模型)和Vicuna-7b(一个微调的7亿参数Llama模型)进行实验。在训练过程中,我们将英语解码器保持冻结,仅更新DNA编码器和投影模型的权重。投影模型在训练开始时从零初始化。
DNA编码器处理DNA序列,并为每个输入标记返回一个嵌入向量,在Nucleotide Transformer模型中,每个标记表示一个6-mer核苷酸。我们记L为DNA序列中的核苷酸数,N为DNA标记的数量(大约)。每个输入的DNA序列如果需要会进行填充,直到最终长度为2,048个标记,约为12 kb。由于DNA编码器的输出嵌入维度可能与英语语言模型的单词嵌入维度不同,我们首先使用一个密集神经网络将每个DNA标记嵌入映射到英语单词维度。在第二阶段,我们使用Perceiver重采样器架构,该架构利用投影的DNA标记嵌入与可学习查询之间的交叉注意力,将N个DNA标记嵌入重采样为K个嵌入向量(补充图2a)。我们已调整此Perceiver重采样器,以包括一个额外的交叉注意力步骤,介于可学习查询和英语问题之间,以从DNA序列中提取依赖于上下文的表示(补充图2b)。
与此同时,英语提示被标记化,并为每个标记生成英语标记嵌入。然后,将K个重采样的DNA嵌入向量插入到英语输入序列中的DNA序列占位符标记位置。如果输入多个DNA序列,则对每个DNA序列分别且独立地应用这些操作。在实践中,我们对K的多个值进行了实验,发现较低的值,如1或4,不能使DNA编码器显著影响冻结的英语解码器的行为。我们发现K=64在英语解码器的输入长度和实践中的性能之间提供了良好的折衷。
在推理过程中,DNA编码器的嵌入仅计算一次。推理是通过自回归的方式进行的,依次预测每个新标记,直到预测到序列结束标记。在生成过程中,英语解码器的关键、查询和值被缓存,以避免计算不必要的操作。我们使用温度采样,温度τ=0.001。
在我们的统一方法中,所有任务都被框定为文本(带有附加DNA输入)到文本的转换,模型生成的输出是一个不受限制的文本序列。对于回归任务,数值值在该序列中生成,并通过交叉熵损失进行优化,而没有任何预定义的结构约束。然而,这种灵活性使得标准回归度量(如MSE)不能直接应用,因为计算MSE需要一个结构化的数字输出格式,并具有指定的占位符。强制这种格式会限制我们方法的通用性和适应性。尽管如此,尽管范围广泛,我们的目标在回归任务中仍取得了竞争性的结果。
整个ChatNT的代码库是在Jax中开发的,使用Haiku进行神经网络实现。所有训练都在8个H100 GPU实例的集群上进行,模型评估可以在单个A100-80GB GPU上完成。所有训练的参数,包括DNA编码器和Perceiver投影的参数,以及优化器累加器和英语解码器的所有冻结参数,均以float32格式存储并更新。
训练
我们在补充表3中描述了ChatNT各部分的所有超参数。ChatNT使用Adam优化器进行训练,学习率为3e−5,其它超参数采用默认设置:β1 = 0.9, β2 = 0.999, ϵ = 1e−8, ϵroot = 0.0。我们使用梯度裁剪值为1,并在批量大小为65,536个标记(相当于256个样本)上累积梯度。我们对每个批次的任务进行了均匀抽样,确保每个批次在每个任务中的样本比例相同。我们在27个任务的数据集上训练模型,训练数据量为20亿个标记(780万样本),在一个由8个H100 GPU实例组成的集群上训练,训练时长为4天。
评估
ChatNT的评估可以在单个A100 GPU上进行,每批次32个样本,并且每个样本生成最多40个标记(每秒13个标记),生成时间为1:40分钟。对于每个任务,我们评估了ChatNT在测试集中的5000个样本上的表现,并报告了相应基准研究中使用的性能度量(补充表1和2)。
基因组学指令数据集
Nucleotide Transformer基准指令
我们为Nucleotide Transformer基准创建了一个指令版本(补充表1)。为了将DNA序列数据集转换为指令数据集,我们为每个任务策划了几十个英语问题和答案,并为每个输入DNA序列抽样一个问题-答案对。在问题中引用输入DNA序列时,我们使用DNA标记占位符<DNA>。答案包含相应输入序列的分类标签。我们将18个二元和多标签分类数据集转换为多样的问题-答案指令,针对每个DNA序列。我们为每个任务提供了训练集和测试集,包含不同的DNA序列以及不同的问题,用以评估模型的表现和英语泛化能力。我们保持了与原始数据集相同的训练和测试集。
新的策划基因组学指令数据集
此处创建的新的基因组学指令数据集包含来自不同研究的27个任务,以英语框定(更多详细信息见补充表2)。它涵盖了与DNA(21个任务)、RNA(3个任务)和蛋白质序列(3个任务)相关的多个调控过程。这些任务来源于多种物种,包括人类、小鼠、果蝇和植物。在所有任务中,包括15个二元分类任务、2个多标签分类任务和10个回归任务。每个任务的训练示例数量从5,500个到300万个不等。有关每个特定任务的数据来源和处理的详细信息,请参见补充信息。
我们按照上述方法将DNA序列数据集转换为指令数据集。答案包含相应输入序列的分类标签或回归分数(最多为小数位)。除了具有单个问题-答案轮次的简单示例和单个序列外,我们还添加了更复杂的示例,其中包含多个轮次的连续问题,这些问题可以是相关的,也可以是无关的,并且交换中问题可以引用多个序列。最终的基因组学指令数据集包含总计6.05亿个DNA标记,即36亿个碱基对,以及2.73亿个英语标记(包括问题和答案)。
我们为每个任务获取了不同DNA序列和不同问题的训练集和测试集,以评估模型的表现和英语泛化能力。
基准数据集
对于27个基因组学任务中的每一个任务,我们将ChatNT的表现与该数据集的最先进方法进行了比较。这些包括卷积神经网络DeepSTARR、ChromTransfer、APARENT2和Saluki,以及基于Nucleotide Transformer、agroNT、DNABERT和ESM2的微调基础模型。我们根据每个任务使用不同的性能度量,遵循相应研究中使用的度量。每个任务的基准方法和性能度量的详细信息可以在补充表2中找到。大多数基准性能度量直接从相关论文中获取。对于ESM2,我们不得不在更新的数据集版本上重新运行它们。
ChatNT预测的校准
我们开发了一种方法来评估和校准ChatNT在二元分类任务中的置信度。
对于给定的二元分类任务,我们从相应任务的测试集中选择N个正面和负面的示例。我们记这些示例分别为和,其中0 ≥ i > N。然后,对于给定的问题x和DNA序列s,我们分别计算模型在正面和负面示例上的平均困惑度。我们将这两个值分别记为和,其中θ表示ChatNT的权重张量。我们按如下方式计算它们:
其中表示答案的第j个标记,返回给定问题、DNA序列和从答案中直到第j个标记的标记的情况下,第j个标记的概率。负面困惑度值以类似方式计算。
这些困惑度值对于正面和负面答案表示模型如何将问题与这些答案对齐的衡量。我们将它们直接解释为logits,并使用softmax变换来计算输入问题的各类别概率。此方法使我们能够从ChatNT中为每个问题示例推导概率。我们对每个任务的1,000个测试示例应用了此方法。
为了校准这些预测,我们首先计算了基于困惑度的概率,使用10,000个训练示例作为我们的校准数据集,并用它们来拟合Platt模型。更具体地,我们使用scikit-learn中的逻辑回归作为校准模型,并使用以下参数进行训练,逆正则化因子C = 0.1,并使用lbfgs求解器。逻辑回归模型学习将ChatNT的基于困惑度的概率映射到更准确的尺度。然后,我们应用该模型来校准上述1,000个测试示例的概率。
作为度量,我们计算了原始基于困惑度的概率和校准后的概率的接收者操作特征曲线下面积(AUROC)和MCC。
归因图计算
我们使用基于梯度的方法计算归因分析的显著性图,使用梯度⨀输入方法。对于每个序列,我们计算模型第一次预测的输出标记(‘是’或‘否’)相对于输入序列的梯度,产生一个位置特定的图,突出显示每个k-mer对模型预测的贡献。
此分析在每个标签的500个测试集序列上进行,任务为剪接供体和剪接受体,并在每个标签的200个序列上进行,任务为TATA启动子。输入序列被标记化为6长度的k-mer,每个标记创建一个one-hot编码向量。这些one-hot编码的标记随后与模型的嵌入矩阵相乘,从而能够计算相对于输入的梯度,而不是它们的离散实体。这些梯度被逐元素相乘,得到每个k-mer在其相应位置的梯度敏感性,按照梯度⨀输入方法。我们取梯度的绝对值,以捕捉每个k-mer对预测输出的贡献大小。
为了总结k-mer的贡献,使用归因图生成每个序列的序列标志,展示每个k-mer的相对重要性。我们使用Logomaker根据归因分数的相对重要性缩放每个k-mer的高度。此外,对于每个归因图,我们识别出归因值最高的五个k-mer并将它们汇总,允许我们通过聚合这些k-mer的出现频率,计算全局k-mer频率。这为我们提供了最具归因值的k-mer及其在所有序列中的频率分布的见解。
最后,我们通过对序列归因图取平均值并选择最大归因位置的k-mer来计算每个任务的序列标志。围绕该k-mer的子序列(包括其左右两侧的区域)被转换为位置权重矩阵,并使用Logomaker可视化为序列标志。