Wannier90文件与参数
Wannier90源码https://github.com/wannier-developers/wannier90/releases/tag/v3.1.0
用法
Wannier90 可以以两种模式运行:
- 后处理模式 Post-processing mode:从文件中读取第一性原理代码计算得到的重叠和投影。我们预计这是使用 wannier90 最常见的方法,详见后文;
-
库模式 Library mode :作为一组库例程,可被第一性原理代码调用,将重叠和投影传递给 wannier90 库例程 wannier90 library routines,并返回与 MLWF 对应的酉变换。如果在第一性原理代码中需要 MLWF,例如在后 LDA 方法中,如 LDA+U 或 SIC,则应使用此方法,详见后文。
wannier90 as a post-processing tool
将 wannier90 用作后处理工具时,代码必须运行两次。
在第一次运行wannier90.x时,要么将输入文件 seedname.win 中的逻辑关键字 postproc_setup 设置为 .true.,要么使用命令行选项 -pp 运行代码。(第一次添加 -pp 指令)。运行代码后会生成文件 seedname.nnkp,该文件提供了构建重叠矩阵元和投影矩阵元
所需的信息。
一旦计算并分别将重叠和投影写入文件 seedname.mmn 和 seedname.amn,将 postproc_setup 设置为 .false. 并第二次运行wannier90.x。输出将写入文件 seedname.wout。(第二次不添加 -pp 指令)
与pwscf (QE)的接口
需要下载并编译 pwscf(即 pw.x 代码)以及后处理接口 pw2wannier90.x
- Run ‘scf’/‘nscf’ calculation with pw.x
pw.x -in scf.in |tee scf.out pw.x -in nscf.in |tee nscf.out - Run wannier90.x
(设置 postproc_setup = .true. 以生成 seedname.nnkp 文件)
- Run pw2wannier90.x
首先读取输入文件(如seedname.pw2wan),该文件定义了基础'scf'计算的prefix和outdir,以及seedname.nnkp文件名,并对从seedname.nnkp读取的直接和倒易晶格矢量与prefix指定文件中定义的矢量进行一致性检查。
pw2wannier90会生成 seedname.mmn 、seedname.amn 和 seedname.eig 文件。
当write_dmn = .true.时(见下文),还会额外创建seedname.dmn和seedname.sym文件。
- Run wannier90.x
(设置 postproc_setup = .false. 以解缠带,定位 MLWF,并使用 MLWF 进行绘图、能带结构、费米面等操作。)
读取 seedname.win 文件,进行Wannier90 主要计算 - Run postw90.x
命令顺序
自洽计算
mpirun -np 32 pw.x -in scf.in > scf.out
↓
非自洽计算
mpirun -np 32 pw.x -in nscf.in > nscf.out
↓
生成seedname.nnkp 文件
wannier90.x -pp znsno3
↓
将QE数据与Wannier90接入
mpirun -np 32 pw2wannier90.x -in znsno3.pw2wan > znsno3.out
↓
进行Wannier主计算
wannier90.x znsno3
or
mpirun -np 32 wannier90.x znsno3
↓
后处理计算,例如计算 shift current
mpirun -np 32 postw90.x znsno3
Wannier能带计算输入文件 seedname.win 示例
# W90
###############
num_bands = 108
num_wann = 108
exclude_bands : 1-102,211-312num_iter = 1000
num_print_cycles = 40dis_win_min = -50.0d0
dis_win_max = 40.0d0
dis_froz_max = 6.5d0
dis_num_iter = 1200
dis_mix_ratio = 1.d0#restart=wannierise
#restart = plot#band_plot
###############
bands_plot : true
bands_num_points : 500
bands_plot_format : gnuplotbegin unit_cell_cart
Ang5.3508901596 0.0000000000 0.0000000000-2.6754450798 4.6340068111 0.00000000000.0000000000 0.0000000000 14.2244462967
end unit_cell_cart#reciprocal lattice unitsBegin Kpoint_Path
G 0.00000 0.00000 0.00000 Z 0.50000 0.50000 0.50000
Z 0.50000 0.50000 0.50000 L 0.00000 0.50000 0.00000
L 0.00000 0.50000 0.00000 G 0.00000 0.00000 0.00000
G 0.00000 0.00000 0.00000 F 0.50000 0.50000 0.00000
End Kpoint_Pathbegin projections
Zn: d
Sn: s;p
O: p
end projectionsbegin atoms_frac
Zn 0.000000000 0.000000000 0.284072012
Zn 0.666666687 0.333333343 0.117405333
Zn 0.666666687 0.333333343 0.617405355
Zn 0.333333343 0.666666687 0.450738668
Zn 0.333333343 0.666666687 0.950738668
Zn 0.000000000 0.000000000 0.784071982
Sn 0.000000000 0.000000000 0.001844000
Sn 0.333333343 0.666666687 0.168510675
Sn 0.666666687 0.333333343 0.335177332
Sn 0.000000000 0.000000000 0.501843989
Sn 0.333333343 0.666666687 0.668510675
Sn 0.666666687 0.333333343 0.835177302
O 0.655207992 0.630455017 0.237561002
O 0.369545013 0.024753001 0.237561002
O 0.975247025 0.344792008 0.237561002
O 0.036211669 0.678125322 0.070894331
O 0.641913652 0.963788331 0.070894331
O 0.321874678 0.358086318 0.070894331
O 0.321874678 0.963788331 0.570894301
O 0.036211669 0.358086318 0.570894301
O 0.641913652 0.678125322 0.570894301
O 0.702878356 0.011458670 0.404227674
O 0.308580339 0.297121674 0.404227674
O 0.988541305 0.691419661 0.404227674
O 0.988541305 0.297121674 0.904227674
O 0.702878356 0.691419661 0.904227674
O 0.308580339 0.011458670 0.904227674
O 0.369545013 0.344792008 0.737560987
O 0.975247025 0.630455017 0.737560987
O 0.655207992 0.024753001 0.737560987
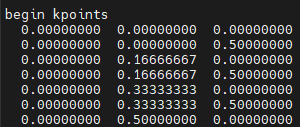
end atoms_fracmp_grid : 6 6 2begin kpoints0.00000000 0.00000000 0.000000000.00000000 0.00000000 0.500000000.00000000 0.16666667 0.000000000.00000000 0.16666667 0.500000000.00000000 0.33333333 0.000000000.00000000 0.33333333 0.500000000.00000000 0.50000000 0.000000000.00000000 0.50000000 0.500000000.00000000 0.66666667 0.000000000.00000000 0.66666667 0.500000000.00000000 0.83333333 0.000000000.00000000 0.83333333 0.500000000.16666667 0.00000000 0.000000000.16666667 0.00000000 0.500000000.16666667 0.16666667 0.000000000.16666667 0.16666667 0.500000000.16666667 0.33333333 0.000000000.16666667 0.33333333 0.500000000.16666667 0.50000000 0.000000000.16666667 0.50000000 0.500000000.16666667 0.66666667 0.000000000.16666667 0.66666667 0.500000000.16666667 0.83333333 0.000000000.16666667 0.83333333 0.500000000.33333333 0.00000000 0.000000000.33333333 0.00000000 0.500000000.33333333 0.16666667 0.000000000.33333333 0.16666667 0.500000000.33333333 0.33333333 0.000000000.33333333 0.33333333 0.500000000.33333333 0.50000000 0.000000000.33333333 0.50000000 0.500000000.33333333 0.66666667 0.000000000.33333333 0.66666667 0.500000000.33333333 0.83333333 0.000000000.33333333 0.83333333 0.500000000.50000000 0.00000000 0.000000000.50000000 0.00000000 0.500000000.50000000 0.16666667 0.000000000.50000000 0.16666667 0.500000000.50000000 0.33333333 0.000000000.50000000 0.33333333 0.500000000.50000000 0.50000000 0.000000000.50000000 0.50000000 0.500000000.50000000 0.66666667 0.000000000.50000000 0.66666667 0.500000000.50000000 0.83333333 0.000000000.50000000 0.83333333 0.500000000.66666667 0.00000000 0.000000000.66666667 0.00000000 0.500000000.66666667 0.16666667 0.000000000.66666667 0.16666667 0.500000000.66666667 0.33333333 0.000000000.66666667 0.33333333 0.500000000.66666667 0.50000000 0.000000000.66666667 0.50000000 0.500000000.66666667 0.66666667 0.000000000.66666667 0.66666667 0.500000000.66666667 0.83333333 0.000000000.66666667 0.83333333 0.500000000.83333333 0.00000000 0.000000000.83333333 0.00000000 0.500000000.83333333 0.16666667 0.000000000.83333333 0.16666667 0.500000000.83333333 0.33333333 0.000000000.83333333 0.33333333 0.500000000.83333333 0.50000000 0.000000000.83333333 0.50000000 0.500000000.83333333 0.66666667 0.000000000.83333333 0.66666667 0.500000000.83333333 0.83333333 0.000000000.83333333 0.83333333 0.50000000
end kpointsQE与Wannier接口输入文件seedname.pw2wan
seedname.pw2wan的示例
&inputppoutdir = './'prefix = 'znsno3_'seedname = 'znsno3'spin_component = 'none'write_mmn = .true.write_amn = .true.write_unk = .false.
/
在 pw2wannier90 输入文件中可以指定一些关键字:
- outdir:输出文件的保存位置。
- prefix :pwscf 计算的前缀
- seedname:Wannier90 计算的seedname
- spin_component:自旋分量。取值为 ‘up’、‘down’ 或 ‘none’(默认值)
- wan_mode:要么是“standalone”(默认值),要么是“library”
- write_unk:设置为 .true. 以在 wannier90 中绘制 Bloch 函数的周期部分。
默认值为 .false.
- reduce_unk:设置为 .true. 可以将 Bloch 函数的文件大小(和分辨率)减少到原来的八分之一,
默认值为 .false.(仅在 write_unk=.true. 时起作用)。
- wvfn_formatted:设置为 .true. 以写入格式化的波函数,
默认值为 .false.(仅在 write_unk=.true. 时起作用)。
- write_amn:如果不需要
默认值为 .true.。
- write_mmn:如果不需要
,则设置为 .false.,
默认值为 .true.。
- write_spn:设置为 .true. 以输出
在布洛赫态之间的矩阵元(仅适用于非共线自旋计算)。默认值为 .false.。
- spn_formatted:设置为 .true. 以将 spn 数据写入格式化文件。
默认值为 .false.(仅在 write_spn=.true. 时起作用)。
- write_uHu:设置为 .true. 以输出矩阵元。
- uHu_formatted:设置为 .true. 以将 uHu 数据作为格式化文件写入。
默认值为 .false.(仅在 write_uHu=.true. 时相关)。
- write_uIu:设置为 .true. 以输出矩阵元。
- uIu_formatted:设置为 .true. 以将 uIu 数据作为格式化文件写入。
默认值为 .false.(仅在 write_uIu=.true. 时相关)。
- write_unkg:设置为 .true. 以写入布洛赫函数周期部分的前几个傅里叶分量。
- write_dmn:设置为 .true. 以构建对称性适应的 Wannier 函数。
默认值为 .false. 。
- read_sym:设置为 .true. 以自定义对称性适应模式中使用的对称操作。当 read_sym = .true. 时,需要额外的输入文件 seedname.sym。
默认值为 .false.(仅在 write_dmn=.true. 时相关)。
不同文件含义
seedname.win 主输入文件
输入文件,具体可以指定的参数见后文的 Wannier90.x 输入文件 [seedname].win 参数 部分
seedname.wout 主输出文件
输出文件
输出的详细程度由输入参数 iprint 控制。数值越高,输出文件提供的细节越多。默认值为 1,表示仅打印最少的信息。
详细解释阅读 Wannier90 user_guide 第80-89页
seedname.nnkp 包含构建重叠&投影所需信息
输出文件
postproc_setup = .true.或者在命令中添加 -pp 时输出,提供了构建重叠矩阵元 和投影矩阵元
所需的信息。用户不应该需要编辑此文件。
seedname.nnkp 中的大部分信息按照,由字符串 begin block_name... end block_name 标记的块分隔的结构排列。
seedname.mmn 重叠矩阵元
输出文件
包含重叠矩阵元
第一行:用户注释,例如日期和时间。
第二行:3个整数:num_bands,num_kpts,nntot。
接下来是 num_kpts × nntot blocks of data:
每个块的第一行:5个整数。
第一个整数指定 k的序号(即给出其在 seedname.win 文件中 k 点列表中的位置序号)。
第 2 到第 5 个整数指定 k + b。
特别是,第 2 个整数指向列表中 k + b 的周期性镜像 k 点,而且特别指出的是实际出现在列表中的那个镜像点。
最后三个整数指定将由第二个整数指定的、且位于第一布里渊区内的 k 点映射到我们需要的实际 k + b 的倒格矢 G,单位为倒易晶格单位。
seedname.amn 投影矩阵元
输出文件
文件 seedname.amn 包含投影矩阵元 。
第一行:用户注释,例如日期和时间。
第二行:3个整数:num_bands、num_kpts、num_wann。
随后是 num_bands × num_wann × num_kpts 行,每行包含 3 个整数和 2 个实数。
前两个整数是能带索引 m 和 n。
第三个整数通过给出其在 seedname.win 文件中 k 点列表的位置序号来指定 k。实数分别是实际 的实部和虚部。
seedname.dmn 包含构建对称性数据
输出文件
文件 seedname.dmn 包含构建对称性适应的 Wannier 函数所需的数据 [7]。如果 site_symmetry = .true.,则需要此文件。
第一行:用户注释,例如日期和时间。
第二行:4个整数:num_bands、nsymmetry、nkptirr、num_kpts。
nsymmetry:对称操作的数量。
nkptirr:不可约k点的数量。
空白行
后跟着num_kpts个整数:完整k点与不可约k点之间的映射。每个k点都与不可约布里渊区中的某个k点相关联。此映射的信息会被写出。每一项对应于全布里渊区中的一个k点,顺序与seedname.win文件中k点列表的出现顺序一致。每一项的(整数)值是该k点在不可约布里渊区中的索引。唯一值的数量等于不可约布里渊区中k点的数量。数据以每行10个值的方式写出。
空白行
后跟着nkptirr个整数:不可约k点的列表。每一项对应于不可约布里渊区中的一个k点。每一项的(整数)值是与seedname.win文件中k点列表相对应的k点索引。这些值应在1到num_kpts之间。数据以每行10个值的方式写出。
空白行
后跟着nkptirr个包含nsymmetry整数数据的块(每个块之间用空白行分隔):通过对不可约k点施加对称操作所得到的k点列表。数据以每行10个值的方式写出。
空白行
nsymmetry × nkptirr 数据块:参考文献[7]中公式(15)的D矩阵信息。每个数据块包含 num_wann × num_wann 行,并且每个块之间用空白行分隔。数据存储在 d_matrix_wann(m,n,isym,ikirr) 中,其中 m, n ∈ [1, num_wann],isym ∈ [1, nsymmetry],ikirr ∈ [1, nkptirr]。元素的顺序是左侧索引比右侧索引变化更快(m:最快,ikirr:最慢)。
空白行
nsymmetry × nkptirr 数据块:参考文献[7]中公式(17)的 d˜ 矩阵信息。每个数据块包含 num_bands × num_bands 行,并且每个块之间由空白行分隔。数据存储在 d_matrix_band(m,n,isym,ikirr) 中,其中 m, n ∈ [1, num_bands],isym ∈ [1, nsymmetry],ikirr ∈ [1, nkptirr]。元素的排列顺序是左侧索引比右侧索引变化更快(m:最快,ikirr:最慢)。
seedname.eig 本征值文件
输出文件
如果 disentanglement、plot_bands、plot_fermi_surface 或 write_hr 中有任何一项为 .true.,则必需。
文件 seedname.eig 包含 Monkhorst-Pack 网格中每个点的 Kohn-Sham 本征值 (以电子伏为单位)。
每行包含两个整数和一个实数。
第一个整数是能带索引
第二个整数表示 seedname.win 文件中 k 点列表中 k 点的序号
实数为本征值。

seedname.sym 对称操作信息
输出文件
如果 read_sym = .true.,则 pw2wannier90.x 需要此附加输入文件;
如果 read_sym = .false.,则此文件由 pw2wannier90.x 输出(仅作参考——后续计算不会使用)。
文件 seedname.sym 包含用于构建对称适应瓦尼尔函数的对称操作信息。如果 read_sym = .false.(默认值),pw2wannier90.x 使用由 pw.x 识别的完整对称性。如果 read_sym = .true.,您可以指定在对称适应模式下使用的对称操作。
第一行:一个整数:nsymmetry(对称操作的数量)
第二行空白
接着:nsymmetry组数据块。每组数据块(由空行分隔)包含四行。每组数据块中的数据顺序如下:

是对称操作的旋转部分(3×3 矩阵),
是以“
”为单位的部分平移
这两组数据均以笛卡尔坐标给出。对称操作作用于点 时的形式为
seedname_band.dat 原始能带数据
输出文件
设置 bands_plot=.TRUE. 来生成;
插值能带结构的原始数据。
seedname_band.gnu GNU能带绘图数据
输出文件
设置 bands_plot=.TRUE. 且 bands_plot_format=gnuplot 来生成
一个用于绘制插值能带结构的 gnuplot 脚本。
seedname_dos.dat 态密度数据
输出文件
在 transport = .TRUE. 时写入。
第一行给出文件创建的日期和时间。
在接下来的行中,左边一列写出以 eV 为单位的能量值,右边一列写出以任意单位表示的态密度。

其他文件
- seedname.chk 检查点文件
输入 & 输出 文件
重新启动计算或进入绘图阶段所需的信息。
如果使用了解纠缠,此文件还包含矩形矩阵
- seedname.r2mn
输出文件
如果 write_r2mn = true,则会写入。
矩阵元素(其中 m 和 n 指代 MLWF)
- seedname_band.agr
输出文件
在 bands_plot=.TRUE. 且 bands_plot_format=xmgrace 时生成;
用于绘制插值能带结构的 grace 文件。
- seedname_band.kpt 插值能带K点数据
输出文件
在 bands_plot=.TRUE. 时生成;
用于插值能带结构的 k 点,单位为倒易晶格矢量。
此文件可用于从第一性原理代码生成对比能带结构。
- seedname.bxsf 费米面数据文件
输出文件
在 fermi_surface_plot=.TRUE. 时生成;
一个适合用 XCrySDen 绘制的费米面图文件。
- seedname_w.xsf 波函数绘图数据,xsf格式
输出文件
在 wannier_plot=.TRUE. 且 wannier_plot_format=xcrysden 时生成。
包含适合用 XCrySDen 或 VMD 绘制的实空间中的第个 WF 的数据。
- seedname_w.cube 波函数绘图数据,cube格式
输出文件
在 wannier_plot=.TRUE. 且 wannier_plot_format=cube 时生成。包含以高斯 cube 格式表示的实空间中的第
- UNKp.s
输入文件
如果 wannier_plot=.TRUE. 则读取,用于绘制 MLWF。
如果 transport_mode=lcr 且 tran_read_ht=.FALSE.,则读取,用于自动化的 lcr 输运计算。
布洛赫态的周期部分表示在规则的实空间网格上,由 k 点 p(从 1 到 num_kpts)和自旋 s(‘1’ 表示 ‘向上’,‘2’ 表示 ‘向下’)索引。
细节阅读 user_guide 第90页
- seedname_centres.xyz 原子结构数据
输出文件
如果 write_xyz=.TRUE.,则会生成一个可由可视化工具(jmol、gopenmol、vmd 等)查看的 xyz 格式原子结构文件。
- seedname_hr.dat 哈密顿矩阵(WF Basis 中的)
输出文件
如果 write_hr=.TRUE. 则生成。
第一行给出文件创建的日期和时间。
第二行说明 Wannier 函数的数量 num_wann。
第三行给出 Wigner-Seitz 网格点的数量 nrpts。
接下来的 nrpts 整数块给出每个 Wigner-Seitz 网格点的简并度,每行 15 个条目。
剩余的行分别包含向量 R 的分量(以晶格矢量
表示)、索引 m 和 n,以及哈密顿矩阵元
在 WF 基下的实部和虚部,例如:
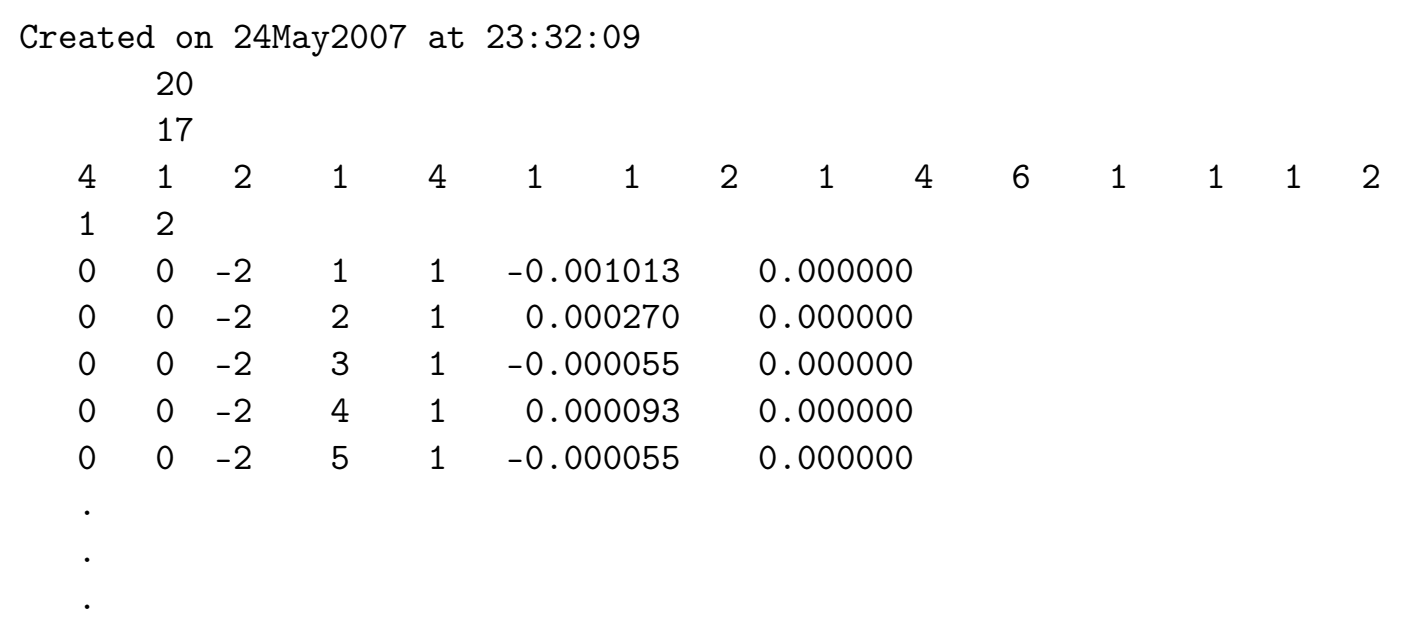
- seedname_r.dat
输出文件
如果 write_rmn 为 true,则会生成
矩阵元(其中 nR 表示单元格 R 中的第 n 个 MLWF)
第一行给出文件创建的日期和时间
第二行说明 Wannier 函数的数量为 num_wann
第三行说明 R 向量的数量为 nrpts
与上面提到的哈密顿矩阵的情况类似,接下来的
- seedname_tb.dat
输出文件
如果 write_tb=.TRUE. 则写入。
此文件实际上是 seedname_hr.dat 和 seedname_r.dat 的组合,再加上晶格矢量。
第一行给出文件创建的日期和时间。
第二至第四行是以埃为单位的晶格矢量

接下来的部分与 seedname_hr.dat 相同。
第五行说明 Wannier 函数的数量为 num_wann。
第六行给出 Wigner-Seitz 网格点的数量 nrpts。
接下来的 nrpts 个整数块给出了每个 Wigner-Seitz 网格点的简并度,每行 15 个条目。
接下来的

最后,最后一部分与 seedname_r.dat 相同。
(第 3 和第 4 列的浮点数分别是的实部和虚部,
第 5 和第 6 列是的实部和虚部,
第 7 和第 8 列是的实部和虚部)

- seedname.bvec
输出文件
如果 write_bvec = true,则会生成该文件。
此文件包含 b-vector 的矩阵元及其权重。
第一行给出文件创建的日期和时间。
第二行说明了 k 点的数量以及每个 k 点的总邻点数 nntot。
然后,所有其他行包含每个 k 点及其每个邻点的 b 向量(x, y, z)坐标和权重。
- seedname_wsvec.dat
输出文件
在 write_hr = true 或 write_rmn = true 或 write_tb = true 时写入。
第一行给出文件创建的日期和时间以及 use_ws_distance 的值。
对于每一对 Wannier 函数(通过向量 R 的分量来标识,R 分隔它们的原胞并用它们的索引来区分),
文件提供:
(i) 周期超胞 T 中使 Wannier 函数从 R 返回到以另一个 Wannier 函数为中心的 Wigner-Seitz 单元的晶格矢量数目;
(ii) 用于实现这一变换的一组超晶格矢量 T。
为了正确地在倒易空间中插值,这些超晶格矢量 T 应加到 R 矢量上,以获得与特定矩阵元(例如 seedname_hr.dat 中的哈密顿量矩阵元)相关的 Wannier 函数的正确中心位置。
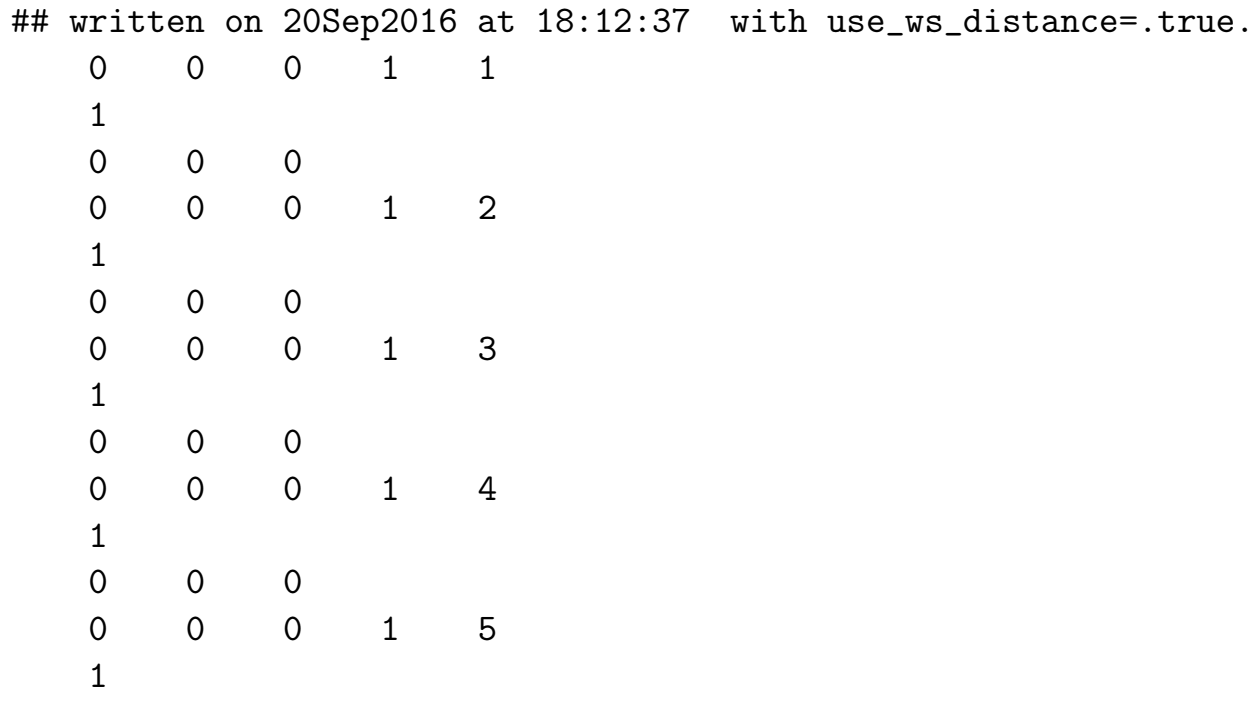
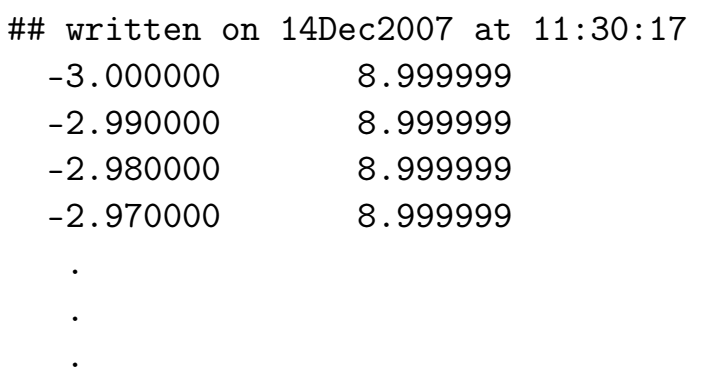
- seedname_qc.dat 量子电导数据
输出文件
在 transport = .TRUE. 时写入。
第一行给出文件创建的日期和时间。
在随后的行中,左边一列写出以 eV 为单位的能量值,右边一列写出以(自旋极化系统为
)为单位的量子电导。
- seedname_htB.dat
输入 & 输出文件
当 transport_mode = bulk 且 tran_read_ht = .TRUE. 时读取,
当 tran_write_ht = .TRUE. 时写入。
首行记录文件创建日期时间,
次行给出 tran_num_bb 数值。
随后各行依次存储:
-主层内 tran_num_bb × tran_num_bb 的
矩阵(m,n索引遍历该层所有tran_num_bb 个WF)
- 新行再次记录 tran_num_bb 数值
- 随后继续列出WF 位于
位于
主层)
(注:矩阵数据采用m索引优先递增的存储格式)
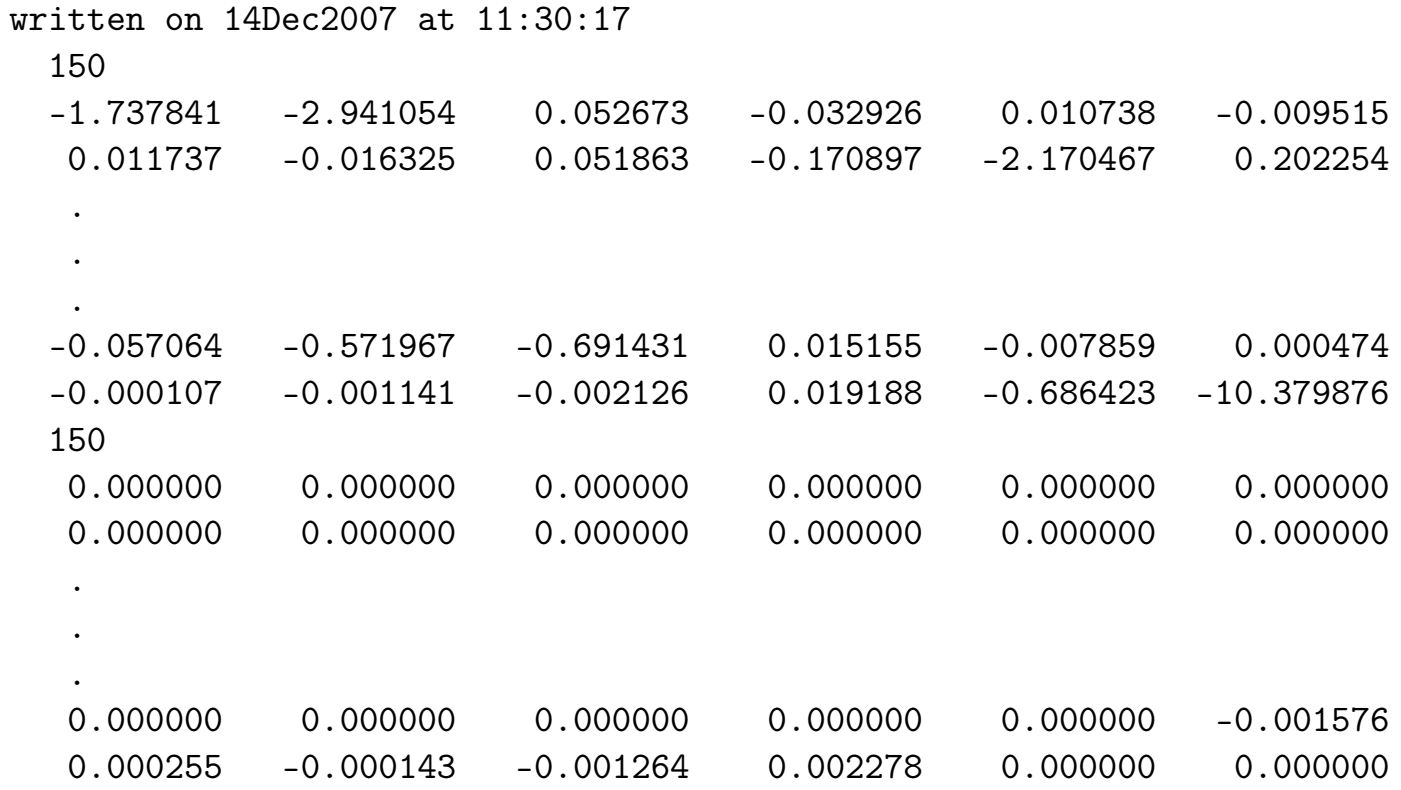
user_guide 第95页
- seedname_htL.dat
输入文件
如果 transport_mode = lcr 且 tran_read_ht = .TRUE.,则读取。
该文件必须以与 seedname_htB.dat 相同的方式写入。

第一行可以是任何你想要的注释。
第二行给出 tran_num_ll。
seedname_htL.dat 中的 tran_num_ll 必须等于 seedname.win 中的 tran_num_ll,否则代码将会停止运行。
- seedname_htR.dat
输入文件
如果 transport_mode = lcr 且 tran_read_ht = .TRUE. 且 tran_use_same_lead = .FALSE.,则读取。
该文件必须以与 seedname_htL.dat 相同的方式写入。
seedname_htR.dat 中的 tran_num_rr 必须与 seedname.win 中的相等。
- seedname_htC.dat
输入文件
如果 transport_mode = lcr 且 tran_read_ht = .TRUE.,则进行读取。
第一行可以是任何你想要的注释。
第二行给出 tran_num_cc。
后续行包含 tran_num_cc×tran_num_cc 的 Hmn 矩阵,其中索引 m 和 n 覆盖中心导体区域内的所有 tran_num_cc 波函数。
seedname_htC.dat 中的 tran_num_cc 必须与 seedname.win 中的相等。

- seedname_htLC.dat
输入文件
如果 transport_mode = lcr 且 tran_read_ht = .TRUE.,则进行读取。
第一行可以是任意你想要的注释。
第二行按给定顺序给出 tran_num_ll 和 tran_num_lc。
后续行包含 tran_num_ll×tran_num_lc 的
索引 m 覆盖与导体区域接触的半无限左引线表面主层中的 tran_num_ll 波函数。
索引 n 覆盖导体区域中与半无限左引线波函数有非可忽略相互作用的 tran_num_lc 波函数。注意,tran_num_lc 可以与 tran_num_cc 不同。

- seedname_htCR.dat
输入文件
如果 transport_mode = lcr 且 tran_read_ht = .TRUE.,则进行读取。
第一行可以是任意注释。
第二行按给定顺序给出 tran_num_cr 和 tran_num_rr。
后续行包含 tran_num_cr×tran_num_rr 的 Hmn 矩阵。
索引 m 覆盖导体区域中与半无限右引线波函数有非可忽略相互作用的 tran_num_cr 波函数。
索引 n 覆盖与导体区域接触的半无限右引线表面主层中的 tran_num_rr 波函数。
注意,tran_num_cr 可以与 tran_num_cc 不同。

- seedname.unkg
输入文件
输入。如果 transport_mode = lcr 且 tran_read_ht = .FALSE.,则进行读取。
第一行是随后打印的 u˜mk(G) 的 G 向量数量。这个数字应始终为 32,因为需要 32 个特定的 u˜mk。
接下来的几行按以下顺序包含以下内容:
带指数 m,G 向量的数量计数器,
G 向量分量的整数系数 a、b、c(其中 G = ab1 + bb2 + cb3),
然后是在 Γ 点对应的 u˜mk(G) 的实部和虚部。
我们注意到,打印 G 向量和 u˜mk(G) 的顺序并不重要,但特定的 G 向量是关键的。
以下示例显示了单个能带所需的完整 u˜mk(G) 集合。请注意所需的 G 向量 (a, b, c)。
user_guide 第97页
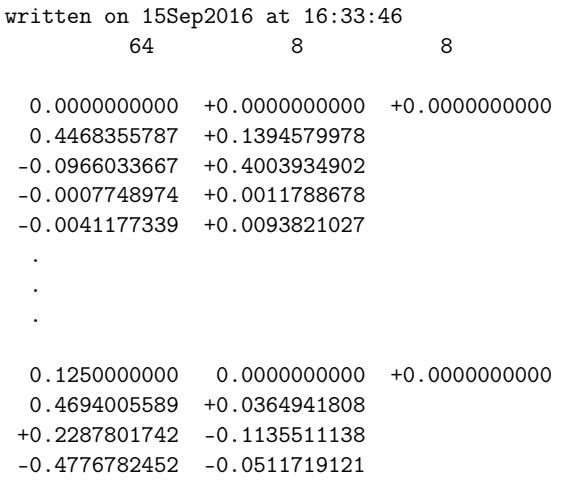
- seedname_u.mat
输出文件
在 write_u_matrices = .TRUE. 时写入。
第一行给出文件创建的日期和时间。
第二行说明了 k 点的数量 num_kpts 和投影函数的数量 num_wann,且 num_wann 出现两次。
第三行为空白行。
接着有 num_kpts 组数据块,每组数据块以一个包含 k 点(用倒易晶格矢量的分数坐标表示)的行开头,
随后是 num_wann * num_wann 行,包含 U(k) 的矩阵元素(实部和虚部)。
矩阵元素按列优先顺序排列(即,先遍历行再遍历列)。
每组数据块之间有一行空行。
- seedname_u_dis.mat
输出文件
在 write_u_matrices = .TRUE. 且解缠功能启用时写入。
第一行给出文件创建的日期和时间。
第二行说明 k 点的数量为 num_kpts,投影函数的数量为 num_bands,以及 num_bands 的数量。
第三行为空白行。
接着有 num_kpts 组数据块,每组数据块以一行包含 k 点(倒易晶格矢量的分数坐标)开头,
随后是 num_wann * num_bands 行,包含 Udis(k) 的矩阵元素(实部和虚部)。
矩阵元素按列优先顺序排列(即先遍历行再遍历列)。
每组数据块之间有一行空行。
Wannier90.x 输入文件 [seedname].win 参数
mpirun -np XXX wannier90.x [-pp] [seedname]
[-pp]:(可选指令)用于命令程序生成后续计算所需要的 重叠积分列表 ,然后退出程序,将相关数据将写入[seedname].nnkp文件。
[seedname]:程序从名为 [seedname].win 的文件中读取输入
Wannier插值能带输入文件案例
# BERRY
###############
berry = true
berry_task=sc
berry_kmesh = 25 25 25
fermi_energy= 5.1158kubo_freq_min= 0.0
kubo_freq_max= 3.0
kubo_freq_step= 0.03
kubo_adpt_smr=truesc_eta=0.040
sc_phase_conv=2
################ W90
###############
num_bands = 108
num_wann = 108
exclude_bands : 1-102,211-312num_iter = 1000
num_print_cycles = 40dis_win_min = -50.0d0
dis_win_max = 40.0d0
dis_froz_max = 6.5d0
dis_num_iter = 1200
dis_mix_ratio = 1.d0#restart=wannierise
#restart = plotbands_plot : true
bands_num_points : 500
bands_plot_format : gnuplotbegin unit_cell_cart
Ang5.3508901596 0.0000000000 0.0000000000-2.6754450798 4.6340068111 0.00000000000.0000000000 0.0000000000 14.2244462967
end unit_cell_cart#reciprocal lattice unitsBegin Kpoint_Path
G 0.00000 0.00000 0.00000 Z 0.50000 0.50000 0.50000
Z 0.50000 0.50000 0.50000 L 0.00000 0.50000 0.00000
L 0.00000 0.50000 0.00000 G 0.00000 0.00000 0.00000
G 0.00000 0.00000 0.00000 F 0.50000 0.50000 0.00000
End Kpoint_Pathbegin projections
Zn: d
Sn: s;p
O: p
end projectionsbegin atoms_frac
Zn 0.000000000 0.000000000 0.284072012
Zn 0.666666687 0.333333343 0.117405333
Zn 0.666666687 0.333333343 0.617405355
Zn 0.333333343 0.666666687 0.450738668
Zn 0.333333343 0.666666687 0.950738668
Zn 0.000000000 0.000000000 0.784071982
Sn 0.000000000 0.000000000 0.001844000
Sn 0.333333343 0.666666687 0.168510675
Sn 0.666666687 0.333333343 0.335177332
Sn 0.000000000 0.000000000 0.501843989
Sn 0.333333343 0.666666687 0.668510675
Sn 0.666666687 0.333333343 0.835177302
O 0.655207992 0.630455017 0.237561002
O 0.369545013 0.024753001 0.237561002
O 0.975247025 0.344792008 0.237561002
O 0.036211669 0.678125322 0.070894331
O 0.641913652 0.963788331 0.070894331
O 0.321874678 0.358086318 0.070894331
O 0.321874678 0.963788331 0.570894301
O 0.036211669 0.358086318 0.570894301
O 0.641913652 0.678125322 0.570894301
O 0.702878356 0.011458670 0.404227674
O 0.308580339 0.297121674 0.404227674
O 0.988541305 0.691419661 0.404227674
O 0.988541305 0.297121674 0.904227674
O 0.702878356 0.691419661 0.904227674
O 0.308580339 0.011458670 0.904227674
O 0.369545013 0.344792008 0.737560987
O 0.975247025 0.630455017 0.737560987
O 0.655207992 0.024753001 0.737560987
end atoms_fracmp_grid : 6 6 2begin kpoints0.00000000 0.00000000 0.000000000.00000000 0.00000000 0.500000000.00000000 0.16666667 0.000000000.00000000 0.16666667 0.500000000.00000000 0.33333333 0.000000000.00000000 0.33333333 0.500000000.00000000 0.50000000 0.000000000.00000000 0.50000000 0.500000000.00000000 0.66666667 0.000000000.00000000 0.66666667 0.500000000.00000000 0.83333333 0.000000000.00000000 0.83333333 0.500000000.16666667 0.00000000 0.000000000.16666667 0.00000000 0.500000000.16666667 0.16666667 0.000000000.16666667 0.16666667 0.500000000.16666667 0.33333333 0.000000000.16666667 0.33333333 0.500000000.16666667 0.50000000 0.000000000.16666667 0.50000000 0.500000000.16666667 0.66666667 0.000000000.16666667 0.66666667 0.500000000.16666667 0.83333333 0.000000000.16666667 0.83333333 0.500000000.33333333 0.00000000 0.000000000.33333333 0.00000000 0.500000000.33333333 0.16666667 0.000000000.33333333 0.16666667 0.500000000.33333333 0.33333333 0.000000000.33333333 0.33333333 0.500000000.33333333 0.50000000 0.000000000.33333333 0.50000000 0.500000000.33333333 0.66666667 0.000000000.33333333 0.66666667 0.500000000.33333333 0.83333333 0.000000000.33333333 0.83333333 0.500000000.50000000 0.00000000 0.000000000.50000000 0.00000000 0.500000000.50000000 0.16666667 0.000000000.50000000 0.16666667 0.500000000.50000000 0.33333333 0.000000000.50000000 0.33333333 0.500000000.50000000 0.50000000 0.000000000.50000000 0.50000000 0.500000000.50000000 0.66666667 0.000000000.50000000 0.66666667 0.500000000.50000000 0.83333333 0.000000000.50000000 0.83333333 0.500000000.66666667 0.00000000 0.000000000.66666667 0.00000000 0.500000000.66666667 0.16666667 0.000000000.66666667 0.16666667 0.500000000.66666667 0.33333333 0.000000000.66666667 0.33333333 0.500000000.66666667 0.50000000 0.000000000.66666667 0.50000000 0.500000000.66666667 0.66666667 0.000000000.66666667 0.66666667 0.500000000.66666667 0.83333333 0.000000000.66666667 0.83333333 0.500000000.83333333 0.00000000 0.000000000.83333333 0.00000000 0.500000000.83333333 0.16666667 0.000000000.83333333 0.16666667 0.500000000.83333333 0.33333333 0.000000000.83333333 0.33333333 0.500000000.83333333 0.50000000 0.000000000.83333333 0.50000000 0.500000000.83333333 0.66666667 0.000000000.83333333 0.66666667 0.500000000.83333333 0.83333333 0.000000000.83333333 0.83333333 0.50000000
end kpoints
任务总体 Job 参数
- num_wann:Wannier函数(WF)的数量(要找到的波函数数量),整数
(我觉得给每个电子分配一个WF应该是合理的吧?
如果您对这方面更了解,欢迎私信我
- num_bands:传递给程序的能带数,整数
Default num_bands=num_wann
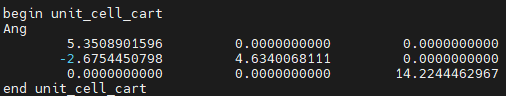
- unit_cell_cart:以笛卡尔坐标 Cartesian 指定 Cell Lattice Vectors
其中[units] 指定了格矢定义所用的单位:要么是 Bohr,要么是 Ang。
如果未指定,则默认为 Ang。
例如

- atoms_cart:以笛卡尔坐标 Cartesian 指定原子位置
第一列是原子符号。
[units] 指定了坐标所用的单位,可以是 Bohr 或 Ang。如果未指定,则默认为 Ang。

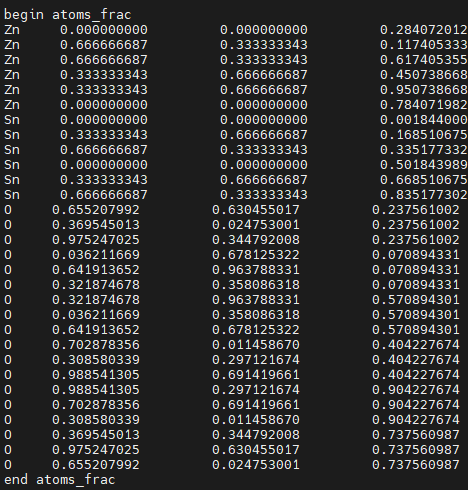
- atoms_frac:原子在分数坐标系 fractional 中位置
第一列是原子符号。
例如

- mp_grid:Monkhorst-Pack K点网格的尺寸,整数
- kpoints:Monkhorst-Pack 网格中的K点列表,实数
每行给出一个 k 点在相对(晶体学)单位下的坐标。
wannier90 提供了一个名为 kmesh.pl 的工具,可以帮助生成 wannier90 所需的显式 k 点列表。
每个 k 点的位置决定了其编号;
第一个 k 点是第 1 号 k 点,
第二个 k 点是第 2 号 k 点,
依此类推。
例如
Wannier90提供了生成给QE与wannier90.x使用的kpoints部分的程序,kmesh.pl
- gamma_only:如果 gamma_only=true,那么 wannier90 会使用一种算法分支来进行解纠缠和局域化计算
- spinors:自旋开关,逻辑值。
如果 spinors=true,那么 wannier90 假设波函数对应于单一占据的旋量态,并且会设置 num_elec_per_state=1。
MV方案需要在均匀的 Monkhorst-Pack k 点网格上定义的 ∇k 的有限差分表达式。向量 {b} 将每个网格点 k 与其最近邻 K 点连接起来.有限差分公式中包含了 层近邻,其中第
层有
个向量。
- shell_list:指定在有限差分公式中使用哪些壳层,整数
shell_list 是一个列出要在有限差分表达式中包含的壳层的向量。如果此关键字缺失,壳层将自动选择。
- search_shells:指定用于定义有限差分公式时,要搜索的壳层数量,整数
指定在尝试确定 B1 条件方程 2.1 的自动解时要搜索的邻近壳层数。在特殊情况下,例如对于非常长且薄的原胞,可能需要比默认值更大的数值。默认值为 36。
- skip_B1_tests:如果设置为 .true.,则不检查 B1 条件方程 2.1。这只有在明确知道为何不应验证 B1 条件时才应使用。此标志的一个典型用法是与 Z2PACK 代码配合使用:http://www.physics.rutgers.edu/z2pack/。
- nnkpts:最近邻K点的列表Explicit list,整数
指定写入 .nnkp 文件的最近邻 k-点。这可以用于明确指定应计算哪些重叠矩阵。

每个最近邻 k + b 由五列整数组成。
第一列表示 k-点编号 nkp。
第二列是近邻的 k-点编号。
最后三列整数表示由前两列指定的 k-点带到 k + b 的倒格矢。
此格式与 .nnkp 文件中的相同,但每个 k-点的邻点数未指定。然而,邻点的数量仍然需要是 k-点数量的倍数。
仅当 postproc_setup = .true. 时可以使用此输入参数,且不建议在完整的 Wannier90 运行中使用。
- kmesh_tol:用于控制两个k点是否属于同一能壳的容差,实数
如果两个 k 点之间的距离小于 kmesh_tol,则它们属于同一个 shell。单位是埃(Ang)。默认值为 0.000001 埃。
- postproc_setup:输出seedname.nnkp文件。逻辑值
如果 postproc_setup=true,那么 wannier 代码将写入 seedname.nnkp 文件并退出。如果用 -pp 选项调用 wannier90,则 postproc_setup 被设置为 true,覆盖 seedname.win 文件中的值。
- exclude_bands:需要从计算中排除的能带列表,整数
用于从重叠矩阵计算中排除某些状态;例如,仅选择价带状态,或忽略半核态。此关键字通过 seedname.nnkp 文件传递给第一性原理代码。
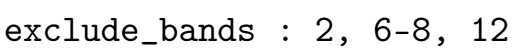
- select_projections:在Wannierisation中使用的投影列表,整数
要在瓦尼尔化过程中包含的投影列表。如果 num_proj 大于 num_wann,此关键字允许使用投影矩阵中的一组子投影。
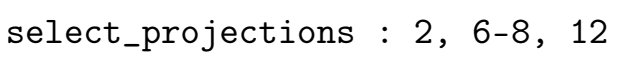
- auto_projections:自动生成初始投影,逻辑值
- restart:从检查点文件重新启动,字符串
如果存在 restart,代码将尝试从 seedname.chk 文件重启计算。该参数的值决定了重启的位置。
– default 从检查文件 seedname.chk 被写入处重启
– wannierise 从 wannierise 程序的开始处重启。
– plot 直接进入绘图阶段
– transport 直接进入 transport 程序
- iprint:输出详细程度级别,整数
输出的详细程度,从 0(“低”),即最少的信息,到 3(“高”),对应于完整的调试输出。默认值为 1。
- length_unit:输出长度的单位制。字符串
输出文件 seedname.wout 中的长度单位。
- Ang(默认)
- Bohr
- wvfn_formatted:从(未)格式化的文件中读取波函数。逻辑值
如果 wvfn_formatted=true,那么波函数将从磁盘读取为格式化(即ASCII)文件;否则它们将被读取为非格式化文件。
通常情况下,非格式化文件更为理想,因为文件占用的磁盘空间更小,并且输入输出速度显著更快。然而,这类文件无法在所有机器架构之间传输,如果需要可传输性(例如,用于测试案例),则应使用格式化文件。此参数的默认值为 false。
- spin:要读取哪个自旋通道,字符串
对于来自自旋极化计算的能带,自旋参数决定读取哪一组能带,向上或向下。此参数的默认值为向上( up )。
- timing_level:控制写入输出文件的计时信息详细程度。整数
确定关于计算的时间信息写入输出文件的数量。值为 1 时产生的信息最少。默认值为 1。
- optimisation:优化级别,整数
代码中使用的优化程度。这是速度和内存之间的一种权衡。正数表示以消耗更多内存为代价,获得最快的执行时间。零或负数表示更小的内存占用,但会增加执行时间。目前,唯一起作用的值是 optimisation<=0(低内存)和 optimisation>0(快速)。默认值为 3。
- translate_home_cell:在写入xyz文件时,将最终的Wannier中心转换到home unit中。逻辑值
确定是否在计算结束时将最终的瓦尼尔中心转换到原胞。这主要用于分子完全位于原胞内的分子系统,并且用户希望编写一个xyz文件(write_xyz=.true.)来将WF中心与结构进行比较。
默认为 false
- write_xyz:将原子位置和最终中心写入到xyz文件格式中。逻辑值
确定是否将原子位置和最终的瓦尼尔中心写入xyz文件,文件名为seedname_centres.xyz,以便后续可视化。
默认为 false
- write_vdw_data:为 w90vdw 可执行文件进一步处理编写数据。逻辑值
确定是否编写 seedname.vdw 文件,以便后续使用 w90vdw 工具(位于分发版的 utility/w90vdw/ 目录下)进行后处理,用于计算范德华能量。布里渊区采样必须仅在伽马点进行。
默认为 false
- write_hr_diag:将哈密顿量在Wannier基底中的对角元素写入到seedname.wout文件中(以电子伏为单位)。逻辑值
投影参数
projections block 定义了一组用于生成单元变换初始猜测的局域函数。此数据将被写入 seedname.nnkp 文件,以供第一性原理代码使用。
如果 guiding_centres=true,则在 Wan-nierisation 过程中将投影中心用作引导中心。
可以设定为

或者按照如下设定

例如

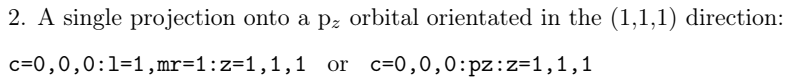

[units]:可选。用于指定给出的投影中心(如果以笛卡尔坐标表示)是以 Ang 为单位还是以 Bohr 为单位。默认值为 Ang。
site:元素符号
ang_mtm:角动量量子态。可以通过 和
来指定,或者使用适当的字符字符串。
和
对应的轨道如下表:


zaxis:可选。
设置形式为 z=1,1,1 会 将 z 轴设置为指向(1,1,1)方向。默认值是 z=0,0,1。
xaxis:可选。
设置形式为 x=1,1,1 会 将 x 轴设置为指向(1,1,1)方向。默认值是 x=1,0,0。
radial:可选。
设置形式为 r=2 指定使用具有一个节点的径向函数(即该角动量的次高伪态)。默认值为 r=1。与不同 r 值相关的径向函数应彼此正交。
zona:可选。
设置形式为 zona=2.0 指定原子轨道径向部分的值(控制径向函数的扩散性)。单位始终为倒数埃。默认值为 zona=1.0。
绘图 Polt 参数
功能:
- 绘制WF
- 绘制插值能带结构
- 绘制费米面
- 输出WF Basis中的哈密顿量
- 输运计算(量子电导和态密度)
#############################################################################
波函数绘制
#############################################################################
- wannier_plot:绘制WF波函数,逻辑值
如果 wannier_plot = true,代码将以 wannier_plot_format 指定的格式输出 Wannier 函数。
默认值为 false。
- wannier_plot_format:用于绘制波函数的文件格式,字符
WF 可以以 XCrySDen (xsf) 格式或 Gaussian cube 格式绘制。
可选参数为:
- xcrysden(默认)
- cube
如果 wannier_plot_format=xsf:代码将在由 wannier_plot_supercell 指定的整个超原胞上输出 WF。
如果 wannier_plot_format=cube:代码会在一个比 wannier_plot_supercell 指定的超原胞小的网格上输出 WF。该网格由 wannier_plot_mode、wannier_plot_radius 和 wannier_plot_scale 决定。
该代码能够为具有非正交晶格矢量的系统输出 Gaussian cube 文件。然而,许多可视化程序(包括XCrySDen)只能处理具有正交晶格矢量的系统的立方文件。
一个能够处理非正交晶格矢量的可视化程序是 VESTA 。
- wannier_plot_list:要绘制的波函数列表,整数
要绘制的波函数列表。波函数按照最小化展布后的 seedname.wout 文件中的编号排列。
默认行为是绘制所有波函数。
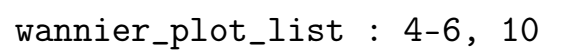
- wannier_plot_supercell:用于绘制波函数的超胞尺寸,整数
代码会在对应于“超原胞”的网格上生成WFs。
如果将 wannier_plot_supercell 设置为整数,那么超原胞的尺寸在所有三个线性方向上都是单元胞尺寸的 wannier_plot_supercell 倍(“本征”单元胞大致保持在中间);
否则,如果提供了三个整数,超原胞的尺寸在第 i 个线性方向上是单元胞尺寸的 wannier_plot_supercell(i) 倍。
默认值为 2。
- wannier_plot_mode:用于绘制波函数的方式,字符
选择绘制 WF 的模式,可以是分子形式或晶体形式。
– crystal (default)
– molecule
如果 wannier_plot_format=cube:
如果 wannier_plot_mode = 分子,那么无论波函数中心位于超晶胞中的哪个位置,立方体的原点都会被移动(仅用于绘图目的,即不对 U 矩阵等进行任何操作),以与用户在 *.win 输入文件中指定的原子位置的质心重合。这些原子位置也会写入 cube 文件,因此在可视化时,波函数会叠加在分子结构上显示。
如果 wannier_plot_mode = 晶体,那么波函数不会发生偏移,而是代码会搜索距离波函数中心在 wannier_plot_scale × wannier_plot_radius 范围内的原子,并将这些原子的坐标写入立方体文件。这样,当 cube 体文件被可视化时,波函数会叠加在其中心附近的原子上显示。
晶体模式可用于分子,分子模式也可用于晶体。
- wannier_plot_radius:波函数截断半径。仅当绘制波函数的文件格式设置为cube时生效。实数。
如果 wannier_plot_format=cube,那么 wannier_plot_radius 是必须容纳在绘制波函数的平行六面体内的球体的半径。wannier_plot_radius 必须大于 0,单位为 Å,
默认值为 3.5
- wannier_plot_scale:cube 文件的缩放参数。实数
如果 wannier_plot_format 设置为 cube 且 wannier_plot_mode 设置为 crystal,
那么程序会查找距离波函数中心半径在 wannier_plot_scale × wannier_plot_radius 范围内的原子,并将这些原子的坐标写入 cube 文件。这样,在可视化 cube 文件时,波函数会叠加在其最近的原子之上。wannier_plot_scale 必须大于 0
该参数无量纲,默认值为 1.0
- wannier_plot_spinor_mode:自旋波函数要绘制的量。字符
如果 spinors = true,那么此参数控制要绘制的量。
对于具有分量的旋量波函数,所绘制的物理量为:
– total (default).
– up. 则当 wannier_plot_spinor_mode = true 时,,否则为
– down. 则当 wannier_plot_spinor_mode = true 时,,否则为
注意:绘制旋量波函数的可视化图像不像绘制标量波函数 scalar WF 那样直观。
虽然标量波函数通常是一个实值函数,但旋量波函数是一个复杂的、具有两个分量的旋量。
wannier90 可以绘制从旋量波函数导出的几种不同量,这应该能帮助你很好地理解该波函数的特性。
- wannier_plot_spinor_phase:绘制自旋波函数时包含“相位”,逻辑值
如果 wannier_plot_spinor_phase = true,在绘制旋量波函数时会考虑相位信息。
#############################################################################
**能带绘制**
#############################################################################
- bands_plot:绘制插值能带结构开关,逻辑值
如果 bands_plot = true,则代码将通过 Wannier 插值计算沿 bands_kpath 定义的 k 空间路径的能带结构,使用 bands_num_points 沿路径的第一段进行计算,并以 bands_plot_format 指定的格式输出结果文件。
默认值为 false。
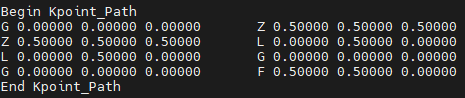
- kpoint_path:插值能带结构的K点路径
定义在 k 空间中计算能带结构的路径。每一行给出路径某一段的起点和终点(带标签)。值是以原始倒格矢 primitive reciprocal lattice vectors 为基准的分数坐标 fractional coordinates 。
例如

- bands_num_points:指定沿k点路径第一段的点数,整数
如果 bands_plot = true,那么能带结构图的第一段的 k 点路径上的点数由 kpoint_path 给出。其他段将具有相同的 k 点密度。
默认值为 100。
- bands_plot_format:指定用于绘制插值能带的文件格式,字符串
用于绘制插值能带结构的格式。此参数的有效选项为:
gnuplot(默认)和 xmgrace。
注意:可以请求同时输出两种格式,例如
- bands_plot_project:指定用于将能带结构投影的波函数,整数
若设置此参数,wannier90将计算这组WF在插值能带结构各点态的贡献。
WF编号遵循seedname.wout文件。
结果写入seedname_band.dat文件,并生成对应的gnuplot脚本seedname_band_proj.dat
例如,要投影到 WFs 2、6、7、8 和 12:

- bands_plot_mode:Slater-Koster型插值或哈密顿量截断,字符
为了沿着 k 点路径插值能带结构,可以使用 Slater-Koster 插值方案,或者在 WF 基底中截断哈密顿矩阵。截断标准由 hr_cutoff 和 dist_cutoff 提供。此参数的有效选项为:
– s-k (default)
– cut
- bands_plot_dim:指定系统维度,整数
系统的维度。如果 bands_plot_dim < 3 且 bands_plot_mode = cut,晶格矢量,其中如果
平行于一维轴(one_dim_axis)指定的任何受限方向,则
,这些将专门用于能带结构插值。
此参数的有效选项为:
– 3 (default)
– 2
– 1
#############################################################################
费米面绘制
#############################################################################
- fermi_surface_plot:绘制费米面开关,逻辑值
如果 fermi_surface_plot = true,
代码将通过 Wannier 插值计算每个方向上带有 fermi_surface_num_points 的规则网格上的本征值。
代码会输出一个 bxsf 格式的文件,该文件可以被 XCrySDen 读取以绘制费米面。
默认值为 false。
- fermi_surface_num_points:指定费米面图中的点数,整数
如果 fermi_surface_plot = true,那么这是用于计算费米面的规则 k 点网格regular k-point grid中的分割数。
默认值为 50。
- fermi_energy:指定费米能级数值
以电子伏特为单位的费米能级。此参数会被写入 bxsf 文件。
如果指定了 fermi_energy,则不应指定 fermi_energy_min、fermi_energy_max 和 fermi_energy_step,反之亦然。
默认值为 0.0。
- fermi_energy_min:费米能级范围的下限
也可以对一系列值范围内的费米能级进行扫描,并为每个重新计算某些量。这是该范围内的最小值(以电子伏特为单位)。
目前,只有 postw90 模块中的 berry 支持扫描费米能级,且仅适用于 berry_task=ahc,morb。对于所有其他需要了解
没有默认值
- fermi_energy_max:费米能级范围的上限
费米能级范围内的最大值。单位为电子伏特。
默认值是 fermi_energy_min+1.0
- fermi_energy_step:在指定范围内提高费米能级的step
在从 fermi_energy_min 扫描到 fermi_energy_max 时,费米能级连续值之间的差值。单位为电子伏特。
默认值为 0.01
- fermi_surface_plot_format:费米面图的文件格式
用于绘制费米面的格式。此参数的有效选项为:xcrysden(默认)
#############################################################################
其他绘图参数控制
#############################################################################
- write_hr:在WF基下写出哈密顿量,逻辑值
如果 write_hr = true,则会将 WF Basis 中的哈密顿矩阵写入到文件 seedname_hr.dat 中。
默认值为 false。
- write_rmn:在WF基下写出位置算符,逻辑值
如果 write_rmn = true,则 WF Basis中的位置算符将被写入文件 seedname_r.dat。
默认值为 false。
- write_bvec:将向量的矩阵元及其权重写入文件。逻辑值
如果 write_bvec = true,则 bvector 的矩阵元及其权重将被写入文件 seedname.bvec。
默认值为 false。
- write_tb:写出晶格矢量、哈密顿量和位置算符在WF基中的表示。逻辑值
如果 write_tb = true,则晶格矢量、哈密顿矩阵以及 WF Basis下的位置算符矩阵将被写入文件 seedname_tb.dat,单位为 Ang 和 eV 。
默认值为 false。
- hr_cutoff:哈密顿量绝对值的截断值
- dist_cutoff:WF之间距离的截断值
- dist_cutoff_mode:计算的WF之间距离的维度,字符串
- translation_centre_frac:
- use_ws_distance:通过最小化WFs之间的距离来改进插值。逻辑值
通过使用一个超晶格的基矢对每个 Wannier 函数进行平移,以最小化它们中心之间的距离,从而改善 k 空间哈密顿量的插值。
该平移取决于 Wannier 函数本身以及它们所属的原胞矢量,即,将函数平移到以
为中心的 Wigner-Seitz 单元内。
若需更详细的解释,请参见第 9 章。
如果设置为 false,则代码会将所有 Wannier 函数置于主单元中,
这是在 wannier90 v2.0.1 之前唯一可行的选择。
默认值为 true(自 v3.0 起默认值已更改)。此功能于 v2.1 引入。
- ws_distance_tol:等效位置距离的绝对容差。实数
在判断两个值和
(如第9章所定义)是否等效时的容差,这两个值表示两个 Wannier 函数之间的最短距离。
如果距离差(以埃为单位)小于 ws_distance_tol,则认为它们是等效的。
默认值为
- ws_search_size:在Born-von Karmann超胞每个方向上的最大扩展范围,用于在Wigner-Seitz胞内搜索点。整数
在 Wigner-Seitz 单元内搜索点时,用于标识超晶格矢量的整数 n、m、l 的最大绝对值(见第9章)。
如果 ws_search_size 被提供为一个单独的整数,则 Born-von Karman 单元在所有三个线性方向上的重复次数相同;
否则,如果提供了三个整数,则第 i 个线性方向上的重复次数为 ws_search_size(i)。
该变量同时在 hamiltonian.F90 和 ws_distance.F90 中使用。
在后一种情况下,其值会增加一,以考虑中心远离原始参考单元的 WFs。
默认值通常足够,但对于拉长的单元可能需要增加。
默认值为 2。
- write_u_matrices:将
和
矩阵写入文件。逻辑值
分别将在 wannierization 结束时获得的
默认值为 false。
控制解纠缠 disentanglement 过程的参数
这些关键词控制参考文献[2]中的解缠程序,即对 的迭代最小化。
如果num_wann < num_bands,此程序将被激活。
- dis_win_min:外层能量窗口的最小值(包含的能量范围)
解缠过程中外层能量窗口的下限。单位为电子伏特。默认值是系统中的最低本征值。
- dis_win_max:外层能量窗口的最大值(包含的能量范围)
解缠过程中外层能量窗口的上限。单位为电子伏特。默认值是给定状态中的最高本征值(即所有状态都包含在解缠过程中)。
- dis_froz_min:内层(冻结)能量窗口的最小值
解缠过程中的内能窗口下限。单位为电子伏特。如果给出了 dis_froz_max,则 dis_froz_min 的默认值为 dis_win_min。
内层(冻结)能量窗口的作用:为了精确保留给定能量范围内(例如,费米能级附近)系统的特性,我们引入了第二个能量窗口。位于这个内部的或“冻结”的能量窗口内的态在最优子空间中保持不变。
- dis_froz_max:内层(冻结)能量窗口的最大值
解缠过程中的内(冻结)能量窗口的上限。如果未指定 dis_froz_max,则不存在冻结态。单位为电子伏特。
- dis_num_iter:用于
最小化的迭代次数,整数
在解缠过程中,用于提取最连接子空间的迭代次数。默认值为 200。
- dis_mix_ratio:在
在解缠过程中,用于收敛的混合参数(参见参考文献[2]的第4-5页)。
0.5 是一个“安全”的选择。
使用 1.0(即不进行混合)通常可以更快收敛,但在某些情况下可能导致 ΩI 的最小化不稳定。
限制:0.0 < dis_mix_ratio ≤ 1.0,
默认值为 0.5。
- dis_conv_tol:用于确定
在解缠过程中,如果在连续迭代中,规范不变扩散的相对变化小于 dis_conv_tol,且这种状态持续 dis_conv_window 次迭代,则认为
默认值为 1.0E-10。
- dis_conv_window:判断
在解缠过程中,如果连续迭代之间的扩散变化量小于 dis_conv_tol 并持续 dis_conv_window 次迭代,则认为最小化过程已收敛。
默认值为 3。
- dis_spheres_num:在 k 空间中执行去纠缠的球体数量,整数
在倒易空间中执行与 k 相关的解缠的球体数量。对于未包含在任何球体中的那些 k 点,不进行解缠。
默认值为 0,这意味着在完整布里渊区的每个 k 点都进行解缠(这是 Wannier90 中的标准模式)。
- dis_spheres_first_wann:Wannier函数的第一个能带的索引,整数
必须被视为 Wannier 函数的第一个能带的索引。
仅在 dis_spheres_num 大于零时使用。
在未进行解缠的 k 点处,从 dis_spheres_first_wann 到 dis_spheres_first_wann + num_wann 的能带用于构建 Wannier 函数。
使用 exclude_bands 排除的能带不应计入。
默认值为 1,即能量最低的能带。
- dis_spheres:去纠缠仅在球体中的中心和半径列表,实数
每一行给出一个 k 点的坐标,该 k 点表示用于 k 依赖解缠的一个球体的中心。这里使用的晶体学单位与 k 点相同。每个 k 点坐标
必须紧接着其对应的球体半径
(以倒易埃 inverse angstrom 为单位,且在同一行)
行数必须等于 dis_spheres_num。

控制Wannier化 wannierisation 的参数
对 进行迭代最小化,这是展宽函数的非规范不变 non-gauge-invariant 部分。
- num_iter:用于
最小化的迭代次数,整数
在最小化过程中的总迭代次数。
如果希望生成投影WF而不是 maximally-localized WFs ( MLWF ),设置 num_iter=0(参见教程中的示例8)
默认值为100。
- num_cg_steps:在
在重置为最速下降之前要采取的共轭梯度步数。
默认值是5。
- conv_window:判断
如果 conv_window > 1,则当
否则,最小化将进行 num_iter 次迭代(默认)。
默认值为 -1。
- conv_tol::用于确定
如果 conv_window > 1,那么这是
默认值是 1.0E-10。
- precond:使用预处理。逻辑值
是否使用预处理来加速展宽的最小化。用于 DFT,可以抑制由于大实空间晶格矢量贡献导致的梯度高频振荡。当优化过程较慢时,特别是在精细网格上,这非常有用。当优化级别小于 3 时,它会使用一种较慢的算法以节省内存。默认值为不使用预处理。
- conv_noise_amp:在最小化过程接近尾声时施加的随机噪声幅度,实数
如果 conv_noise_amp > 0,在达到收敛(如上所述)后,会向搜索方向添加一些随机噪声 f,并继续最小化过程,直到再次达到收敛。如果得到的 Ω 值与之前相同,则认为计算已收敛。如果不是,则再次添加随机噪声,并重复此过程,最多重复 conv_noise_num 次。conv_noise_amp 是添加到搜索方向的随机噪声 f 的幅度:0 < |f| < conv_noise_amp。
此功能需要 conv_window > 1。
如果未指定 conv_window,则默认设置为 5。
如果 conv_noise_amp ≤ 0,则不添加任何噪声(默认)。
默认值为 -1.0。
- conv_noise_num:随机噪声施加的次数,整数
如果 conv_noise_amp > 0,那么这是在最小化过程中添加随机噪声的次数。
默认值是 3。
- num_dump_cycles:控制检查点的频率,整数
每 num_dump_cycles 次迭代写入足够的信息以便进行重启。
默认值为 100。
- num_print_cycles:控制打印频率,整数
每 num_print_cycles 次迭代将数据写入主输出文件 seedname.wout。
默认值为 1
- write_r2mn:将WF之间的
矩阵元素写入文件。逻辑值
如果 write_r2mn = true,则在 Wannier 化程序结束时,会将矩阵元素 hm|r2|ni(其中 m 和 n 指代波函数)写入到文件 seedname.r2mn 中。
默认值为 false。
- guiding_centres:使用guiding centres,逻辑值
在最小化过程中使用引导中心,以避免局部最小值。
wannier90 使用扩展功能的对数定义。由于我们要对复数参数取对数,算法可能会对分支切割做出不一致的选择。这表现为具有较大扩展的复数波函数。通过使用引导中心,代码将尝试做出一致的分支切割选择。经验表明,将引导中心设置为 true 可以避免此问题,并且这样做不会引起任何问题。因此,我们建议在可能的情况下将 guiding_centres 设置为 true(仅在未定义显式投影块时才不可能)。
默认值为 false。
- num_guide_cycles:guiding centres的频率
如果将 guiding_centres 设置为 true,则引导中心每 num_guide_cycles 使用一次。
默认值为 1。
- num_no_guide_iter:使用 guiding centres的迭代次数
如果引导中心被设置为 true,则只有在完成 num_no_guide_iter 次最小化迭代后才会使用引导中心。
默认值为 0。
- trial_step:在最小化
在用于扩散函数最小化的线搜索最小化过程中,抛物线拟合的试探步长值。
不能与 fixed_step(见下文)同时使用。
如果最小化过程无法收敛,尝试减小 trial_step 的值以获得更精确的线搜索。
默认值为 2.0。
- fixed_step:在最小化
若在输入文件中指定该参数值,则在每次扩展函数最小化迭代时,将采用固定步长(fixed_step)替代抛物线线性搜索。
当线性搜索法无法收敛时,强制固定步长可能改善稳定性
- use_bloch_phases :使用相位进行初始投影,逻辑值
确定是否将布洛赫函数用作投影的初始猜测。
只有在 disentanglement = false 时才能使用。
默认值为 false。
- site_symmetry:构建对称性适应的瓦尼尔函数,逻辑值
构建对称性适应的瓦尼尔函数。
不能与 inner (frozen) energy window 同时使用。
默认值为 false。
- symmetrize_eps:在对称性适应模式下使用的收敛容差,实数
用于检查单位矩阵 U(k) 是否满足对称性条件(参考文献[7]中的公式(19))的收敛阈值。另见参考文献[7]中的公式(29)。
当 site_symmetry = .true 时使用。
默认值为 1.0E-3。
- slwf_num:选择性局域化的目标WF数量,整数
选择性局域化的目标瓦尼尔函数的数量。
这些函数是通过仅针对 slwf_num < num_wann 函数的子集的自由度最小化扩展泛函获得的。在收敛时,目标瓦尼尔函数将具有最小的累积扩展,而其余的 num_wann − slwf_num 函数保持未优化状态。目标瓦尼尔函数的初始猜测由投影块中的前 slwf_num 轨道给出。
如果 slwf_num = num_wann,则不执行选择性最小化。在这种情况下,wannier90 将简单地生成一组包含 num_wann 个 MLWF 的集合。
默认值为 num_wann。
- slwf_constrain:是否约束目标WFs的中心,逻辑值
如果 slwf_constrain=true,那么目标瓦尼尔函数的中心将被限制在投影块中前 slwf_num 个轨道的中心,或者限制在 slwf_centres 块中指定的新位置(见第 2.8.22 节)。在这种情况下,如参考文献[8]所述,使用带有约束项的修正扩展泛函。
默认值为 false。
- slwf_lambda:用于约束目标WF的拉格朗日乘子的值,实数
- slwf_centres:目标WF将被约束的中心
如果 slwf_constrain=true,那么默认情况下,slwf_num 个目标瓦尼尔函数中心所约束的中心由投影块的前 slwf_num 行给出。
可选地,可以使用 slwf_centres 块为部分或全部 slwf_num 个目标瓦尼尔函数定义替代的目标中心。

第一行将目标WF编号2(按投影块中WF的顺序定义)的中心约束设置为分数坐标中的 (0.0, 0.0, 0.0)。
第二行将目标WF编号4(按投影块中WF的顺序定义)的中心约束设置为分数坐标中的 (0.25, 0.0, 0.0)。
所有其他目标Wannier函数的中心仍保持为投影块中对应行给出的中心。
控制传输 transport 的参数
- transport:计算量子电导和态密度,逻辑值
如果 transport = true,代码将计算一维系统的量子电导和态密度。
结果将分别写入文件 seedname_qc.dat 和 seedname_dos.dat。
由于这两个量都是能量的函数,它们将从 tran_win_min 评估到 tran_win_max,间隔为 tran_energy_step。
此参数的默认值为 false。
- transport_mode:体材料或左引线_导体_右引线计算,字符串
可选:
– bulk (default)
– lcr
如果 transport_mode = bulk,将为一个完全周期性的一维系统计算量子电导和态密度。在这种情况下,输运部分可以使用 wannier90 生成的 WF 基下的哈密顿矩阵,也可以使用外部文件 seedname_htB.dat 提供的哈密顿矩阵。
如果 transport_mode = lcr,将为一个系统计算量子电导和态密度,该系统中半无限的左引线和右引线通过中心导体区域连接。在这种情况下,输运部分将独立于去纠缠和瓦尼尔化过程运行。该方法的详细内容参见参考文献[9]。
如果 tran_read_ht = true,则哈密顿矩阵必须由五个外部文件提供:seedname_htL.dat、seedname_htLC.dat、seedname_htC.dat、seedname_htCR.dat、seedname_htR.dat。
如果 tran_read_ht = false,则哈密顿矩阵会自动找到,前提是超胞符合第 7.3 节中概述的条件。
- tran_read_ht:读取用于输运计算的哈密顿量。逻辑值
- tran_win_min:transport calculation 中能量窗口的下限
输运计算中能量窗口的下限。单位为电子伏特。
默认值为 -3.0
- tran_win_max:transport calculation 中能量窗口的上限
输运计算中能量窗口的上限。单位为电子伏特。
默认值为 3.0
- tran_energy_step:能量值的采样间隔。实数
从 tran_win_min 到 tran_win_max 的能量值采样间隔。单位为电子伏特。
默认值为 0.01
- fermi_energy:费米能级。实数
费米能级,单位为电子伏特。
量子电导和态密度数据的能量轴将以此数值刚性平移。
默认值为 0.0。
- tran_num_bb:bulk 哈密顿量的尺寸,整数
体哈密顿矩阵的大小。
这个数值等于一个主层中的 WFs 数量。
一维系统可以被看作是主层的数组,定义方式为某个主层内的局域基函数仅与最近邻主层内的基函数相互作用。
在 wannier90 中,一个主层将是原胞的整数倍,其大小由 hr_cutoff 和/或 dist_cutoff 决定。
当采用 WFs 作为局域基函数时,判断标准较为任意,取决于用户的选择。 - 默认值为 0。
- tran_num_ll:left-lead 哈密顿量的尺寸,整数
左引线 left-lead 哈密顿矩阵的大小。
如果 transport_mode = lcr 且 tran_read_ht = false,那么 tran_num_ll 是主层中 Wannier 函数的数量。
默认值为 0。
- tran_num_rr:right-lead 哈密顿量的尺寸,整数
右引线 right-lead 哈密顿矩阵的大小。
默认值为 0。
- tran_num_cc:conductor 哈密顿量的尺寸,整数
导体 conductor 哈密顿矩阵的大小。
默认值为 0。
- tran_num_lc:left-lead_conductor 哈密顿量的列数,整数
左引线-导体 left-lead_conductor 哈密顿矩阵的列数。行数必须等于 tran_num_ll。
默认值为 0。
- tran_num_cr:conductor_right-lead 哈密顿量的行数,整数
导体_右引线 conductor_right-lead 哈密顿矩阵的行数。列数必须等于 tran_num_rr。
默认值为 0。
- tran_num_cell_ll:left-lead 中PL的 unit cells 数量,整数
左引线 left lead 一个主层 principal layer 中的单元格数量 Number of unit cells 。如果 transport_mode = lcr 且 tran_read_ht = false,则使用。
默认值为 0。
- tran_num_cell_rr:right-lead 中PL的 unit cells 数量,整数
右引线一个主层中的单元格数量。目前未使用。
默认值为 0。
- tran_num_bandc:band-diagonal conductor 哈密顿量的 Half-bandwidth+1,整数
能带对角线导体 band-diagonal conductor 哈密顿矩阵的 Half-bandwidth+1 。
从 seedname_htC.dat 读取的中心导体部分的哈密顿矩阵,在 tran_num_cc 非常大时将呈现对角占优。tran_num_bandc 用于构建一个紧凑矩阵,该矩阵包含完整导体哈密顿矩阵的非零带对角部分。
仅当 tran_num_bandc 大于 tran_num_lc 和 tran_num_cr 时,设置此参数才有意义。
默认值为 0。
- tran_write_ht:写出用于输运计算的哈密顿量。逻辑值
如果 tran_write_ht = true,则用于传输计算格式化的哈密顿矩阵将被写入到文件 seedname_htB.dat 中。
默认值为 false。
- tran_read_ht:
如果 tran_write_ht = true,则用于传输计算格式化的哈密顿矩阵将从参数 transport_mode 中描述的一组文件中读取。
设置 tran_write_ht = false 以执行自动的 lcr 计算(见第 7.3 节)。
默认值为 false。
- tran_use_same_lead:设置 Left 与 right leads 是否相同,逻辑值
如果 tran_use_same_lead = true,则左引线和右引线相同。
在这种情况下,不需要 seedname_htR.dat 文件。
默认值为 true。
- tran_group_threshold:决定 WFs 分组的距离。实数
用于根据中心位置对 Wannier 函数进行分组和排序。
分组中的 Wannier 函数在 x、y 和 z 方向上彼此之间的距离不超过 tran_group_threshold。
单位为 Å。
默认值为 0.15。
- hr_cutoff:哈密顿量绝对值的截断。
哈密顿量在WF Basis 中最小矩阵元的绝对值。
如果> hr_cutoff,则矩阵元
否则,它将被视为不重要而被舍弃。单位为电子伏特 (eV)。
默认值为 0.0
- dist_cutoff:波函数之间距离的截断
两个WF之间保留并在能带插值(当 bands_plot_mode = cut 时)或输运计算中使用的哈密顿量矩阵元的最大距离。单位为Å。
默认值为1000.0。
- dist_cutoff_mode:计算波函数之间距离的维度,字符串
计算两个WF之间距离的维度。
连接两个WF的向量可以投影到一条直线(one_dim)或一个平面(two_dim)。
计算投影向量的大小,并应用 dist_cutoff。
当使用 one_dim 或 two_dim 时,必须给出 one_dim_axis 以指定延伸或限制的方向。
可选参数:
– three_dim (default)
– two_dim
– one_dim
- one_dim_axis:一维系统的延伸方向,字符串
一维系统的延伸方向或二维系统的限制方向。此方向必须与笛卡尔坐标轴Cartesian axes之一平行。
可选参数:
– x
– y
– z
- translation_centre_frac:unit cell 的中心,最终波函数将转换至此。实数
最终 Wannier 中心所平移至的原胞中心。数值以晶格矢量为基准的分数坐标fractional coordinates表示。
默认值为(0.0, 0.0, 0.0)。
postw90.x 功能
postw9o.x 使用与 wannier90.x 相同的 seedname.win 输入文件。因此,必须使用第 2.2 节中所述的相同语法将 postw90.x 的输入关键字添加到此文件中。
wannier90.x 会检查输入文件的语法是否正确,但会忽略仅与 postw90.x 模块相关的
标志的值,因此可以放心地在包含 postw90.x 标志的文件上运行 wannier90.x。
因此可以直接将 postw90 输入关键词添加到用于获取 Wannier 函数的 seedname.win 文件中。让两个程序使用同一个输入文件 .win
postw90.x 当前可用的模块有:
- dos:态密度(DOS)、投影态密度(PDOS)、净自旋等的计算
- kpath:在布里渊区(BZ)内沿分段线性路径计算 k 空间量,例如能带、贝里曲率以及自旋霍尔电导率的贝里曲率类似项
- kslice:在布里渊区(BZ)的平面切片上计算 k 空间量
- berry:与贝里曲率、贝里联络以及自旋霍尔电导率的贝里曲率类似项的布里渊区积分相关的性质计算,包括反常霍尔电导率、轨道磁化率、光学电导率、非线性位移电流和自旋霍尔电导率
- gyrotropic 旋光:旋光特性的计算,包括自然旋光和电流诱导旋光,以及电流诱导磁化
- BoltzWann:使用半经典玻尔兹曼输运方程计算体材料的电子输运性质。在 Wannier 基下计算玻尔兹曼输运系数
- geninterp(通用能带插值):在通用的 k 点列表上计算能带能量(及能带导数)
全局参数
- kmesh
在 postw90.x 中使用的插值网格的尺寸。
切勿将其与 mp_grid 输入标志相混淆!
如果给定了三个整数 l、m、n,则在包括 Γ 点的均匀 l×m×n 网格上对由三个原胞平移向量所界定的倒易空间单元进行采样。如果仅给定一个整数 m,则使用 m×m×m 网格。
如果使用需要 k 网格的模块,则必须定义 kmesh_spacing 或 kmesh。
- kmesh_spacing
指定插值网格的另一种方式。此标志定义了 k 空间中沿三个方向相邻 k 点之间的最小距离。
单位为 1/Å
- adpt_smr
确定是否使用自适应方案来展宽态密度(DOS)和定义在能量轴上的类似量。如果为真,则展宽的值由标志 adpt_smr_fac 控制。
默认值为 true 。
- adpt_smr_fac
用于确定第 n 个带在 k 点对谱性质(态密度,...)的贡献时,所使用的展宽delta函数的宽度计算如下:
其中是能量本征值,无量纲因子
由 adpt_smr_fac 给出。
被取为沿三个倒格矢 b1、b2 和 b3 的网格间距中最大的一个。
如果计算得到的 ηnk 超过 adpt_smr_max,则使用后者作为值。
默认值为
- adpt_smr_max
默认值为1.0
- smr_type
定义了在计算态密度(DOS)和能量轴上类似量时所使用的展宽 delta 函数的解析形式。
可选项有:
- gauss:高斯展宽
- m-pN:第 N 阶 Methfessel-Paxton 函数的导数(N ≥ 0)
- m-v / cold:Marzari–Vanderbilt 冷平滑函数的导数
- f-d:费米-狄拉克分布函数的导数
默认值为 gauss
- smr_fixed_en_width
用于态密度(DOS)的平滑函数的能量宽度。
仅在“adpt_smr”为 false 时使用。单位是电子伏特(eV)。
“能量宽度”的默认值为 0 eV。
请注意,如果宽度小于能量步长的两倍(例如,态密度模块中的“dos_energy_step”),则态密度将不会被平滑(因此,默认情况下,当“adpt_smr”设置为 false 时,属性不会被平滑)。
- num_elec_per_state
每个状态的电子数。它只能取为 1 或 2 。
如果 spinors=true,默认值为 1,否则默认值为 2。
- num_valence_bands
系统的价带数量。
在不同模块中使用,也用于剪刀修正。无默认值。
- spin_decomp
如果为真,则会在某些输出文件中添加额外的列
(例如,用于密度泛函模块的 seedname-dos.dat 文件,以及类似的 berry 和 BoltzWann 模块的文件)。
对于 dos 和 BoltzWann 模块,
会生成两列额外的列,包含自旋计算中所需性质(例如总 DOS 或轨道投影 DOS)分解为上自旋和下自旋部分的结果(相对于由输入变量 spin_axis_polar 和 spin_axis_azimuth 定义的量化轴)。
对于 berry_task = kubo 的 berry 模块,在 seedname-jdos.dat 文件中添加三列额外的列,
包含 JDOS 分解为 up → up 、down → down 以及自旋翻转跃迁 spin-flip transitions 的部分。
同样地,在存储复数光学电导率 complex optical conductivity 的数据文件
seedname-kubo*.dat 中添加六列额外的列。
输入时必须存在文件 seedname.spn。
此外,如果此变量设置为 true,则需要 num_elec_per_state = 1。
默认值为 false。
- spin_axis_polar
自旋量化轴的极角。
单位为 degrees。默认值为 0。
- spin_axis_azimuth
自旋量化轴的方位角。
单位为 degrees。默认值为 0
- spin_moment
确定是否计算自旋矩 spin moment。
默认值为 false。
- uHu_formatted
如果 uHu_formatted=true,则 uHu 矩阵元将从磁盘读取为格式化(即 ASCII)文件;
否则,它们将被读取为非格式化文件。
此参数的默认值为 false。
- spn_formatted
如果 spn_formatted=true,则自旋矩阵元将从磁盘读取为格式化(即 ASCII)文件;
否则,它们将被读取为非格式化文件。
通常更倾向于读取非格式化文件,因为文件占用的磁盘空间更少,并且输入输出速度显著更快。
但是,这样的文件并不能在所有机器架构之间进行传输,如果需要可传输性(即,用于测试案例时),应使用格式化文件。
默认值为 false
- berry_curv_unit
在输入(在 berry_curv_adpt_kmesh_thresh 中)或写入文件(当 kpath_task=curv 或 kpath_task=shc 或 kslice_task=curv 或 kslice_task=shc 时)时,
用于指定贝里曲率的单位。
可选参数:
- ang2:
- bohr2:(atomic units)
默认值为 ang2
**berry 模块参数**
- berry
确定是否进入贝里例程。
默认值为 false。
- berry_task
在 berry=true 时要计算的量。
此参数的有效选项为:
- kubo:复光学电导率 optical conductivity 和联合态密度 joint density of states。输出文件:
seedname-kubo-S_{xx,yy,zz,xy,xz,yz}.dat 对称电导率
第一列:光学频率,单位为电子伏特(
)。
第二列和第三列:对称电导率的实部和虚部,
单位为西门子每厘米
如果 spin_decomp = true,则会额外出现六列。
seedname-kubo-A_{yz,zx,xy}.dat 反对称电导率
第一列:光学频率 ¯hω,单位为
第二列和第三列:反对称电导率的实部和虚部,
单位为西门子每厘米
如果 spin_decomp = true,则会额外出现六列。
seedname-jdos.dat JDOS计算
第一列:导带()与价带(
)状态之间的能量差
,
单位为
第二列:态密度
(每单位晶胞每单位能量范围内的态数量,单位为)。
如果 spin_decomp = true,则会额外出现三列。
- ahc:反常霍尔电导,单位为 S/cm。
计算三个独立分量、
和
输出文件:
seedname-ahc-fermiscan.dat
如果通过 fermi_energy_min 和 fermi_energy_max
指定了一个费米能级范围,则会生成此文件。
第一列包含费米能级
随后的三列包含的值。
如果仅给定单一费米能级值,
则反常霍尔电导仅打印在 seedname.wpout 中。
- morb:轨道磁化强度,单位为bohr magnetons per cell
输出文件:
seedname-morb-fermiscan.dat
如果通过 fermi_energy_min 和 fermi_energy_max 指定了费米能级范围,
则会生成此文件。
第一列包含费米能级
随后的三列包含的值。
如果仅给定单一费米能级值,则仅打印在 seedname.wpout 中。
- shc:自旋霍尔电导率(Spin Hall conductivity,SHC),单位为
输出文件:
seedname-shc-fermiscan.dat
如果通过 fermi_energy_min 和 fermi_energy_max 指定了费米能级范围,
则会生成此文件。
第一列是列表中的条目数,
第二列包含费米能级
最后一列包含的值。
如果仅给定单一费米能级值,
该文件将包含在此特定能量下的自旋霍尔电导率。
seedname-shc-freqscan.dat
如果通过 kubo_freq_min 和 kubo_freq_max 指定了频率范围,
则会生成此文件
第一列是列表中条目的编号,
第二列包含频率
接下来的两列分别包含交流自旋霍尔电导率的
实部
虚部![\Im[\sigma_{\alpha\beta}^{\mathrm{spin}\gamma}(\omega)]](https://latex.csdn.net/eq?%5CIm%5B%5Csigma_%7B%5Calpha%5Cbeta%7D%5E%7B%5Cmathrm%7Bspin%7D%5Cgamma%7D%28%5Comega%29%5D)
- berry_kmesh
覆盖全局变量中的 kmesh 的值,在 postw90.x 中使用的新的插值网格的尺寸。or
- berry_kmesh_spacing
覆盖全局变量中的 kmesh_spacing 的值,在 postw90.x 中使用的新的插值网格的尺寸。
- berry_curv_adpt_kmesh
如果给定正整数 n 并且 berry_task=ahc [或者 berry_task=shc] ,
那么在均匀网格(由 berry_kmesh 或者 berry_kmesh_spacing 定义)中,对于那些超过 berry_curv_adpt_kmesh_thresh 指定阈值的 k 空间贝里曲率[k 空间自旋霍尔相关的贝里曲率类项] 的点,会放置一个 n × n × n 的网格。
这可以用于在贝里曲率[自旋霍尔相关的贝里曲率类项] 的尖峰周围加密布里渊区积分网格。
默认值为 1。
- berry_curv_adpt_kmesh_thresh
触发自适应网格细化的贝里曲率[自旋霍尔相关的贝里曲率类项] 的大小(以 berry_curv_unit 为单位),
当 berry_task=ahc[berry_task=shc] 时使用。
默认值为 100.0。
- kubo_freq_min
计算光学电导率、联合密度态和交流自旋霍尔电导的频率范围下限。
单位为
- kubo_freq_max
计算 光学电导率、联合密度态和交流自旋霍尔电导 的频率范围上限。
单位为
如果指定了内部能量窗口 dis_froz window,则默认值为 dis_froz_max 减去 fermi_energy 加上 0.6667。
否则,它是存储在 seedname.eig 文件中的最大能量本征值与最小能量本征值之差,再加上 0.6667。
- kubo_freq_step
介于 kubo_freq_min 和 kubo_freq_max 之间的光学频率连续值的差值。
单位为
- kubo_eigval_max
在计算 光学电导率、JDOS 和交流自旋霍尔电导 时要包含的本征态的最大能量本征值。
单位为
如果指定了内部能量窗口,则默认值是内部能量窗口的上限加上 0.6667。
否则,它是存储在 seedname.eig 中的最大能量本征值加上 0.6667。
- kubo_adpt_smr
覆盖全局变量中的 adpt_smr
- kubo_adpt_smr_fac
覆盖全局变量中的 adpt_smr_fac
- kubo_adpt_smr_max
覆盖全局变量中的 adpt_smr_max
- kubo_smr_type
覆盖全局变量中的 smr_type
- kubo_smr_fixed_en_width
覆盖全局变量中的 smr_fixed_en_width
- sc_eta
用于展宽分母中能量差的宽度 η
上述内容在移位电流计算中是必需的,目的是为了避免在对虚拟态求和时,由于近简并引起的数值问题。
单位为
默认值为 0.4。
- sc_phase_conv
布洛赫态在移位电流计算中展开的约定。它只能取一或二这两个值。
我们遵循参考文献[11]中的约定:
1:在相位因子中包含 Wannier 中心(即所谓的紧束缚约定):
2:不在相位因子中包含 Wannier 中心(通常的 Wannier90 约定):
该约定不会影响完整的移位电流矩阵元,但会影响构成它的内部组件的权重(见参考文献[12])。
默认值为 1。
- sc_w_thr
用于加速移位电流计算中频率积分的参数。
它确定了频率阈值(一个乘到展宽上的的因子),
超过该阈值后,delta 函数被视为零。
默认值为 5.0
- shc_freq_scan
确定是计算自旋霍尔电导率的频率扫描(即交流 SHC)还是费米能级扫描(即直流 SHC)。默认值为 false,这意味着计算直流 SHC。
- shc_alpha
自旋霍尔电导率的
可选的值为 1、2 和 3,分别表示 x、y 和 z 方向。
默认值为 1。
- shc_beta
自旋霍尔电导率索引,即外加电场的方向。
可选的值为 1、2 和 3,分别表示 x、y 和 z 方向。
默认值为 2。
- shc_gamma
自旋霍尔电导率索引,即自旋电流的自旋方向。
可选的值为 1、2 和 3,分别表示 x、y 和 z 方向。
默认值为 3。
如果 shc_alpha、shc_beta 和 shc_gamma 都设置为默认值,则计算。
- shc_bandshift
将所有导带平移一个给定的量(由 shc_bandshift_energyshift 定义)。
注意:此标志与全局的 scissors_shift 标志略有不同:
使用 shc_bandshift 时,在插值后会应用精确的刚性平移;
而 scissors_shift 则是在插值前应用平移。
因此,结果可能会略有不同(这也是我们提供两种可能性的原因)。
另外需要注意的是,
使用 scissors_shift 时,你需要提供价带的数量 num_valence_bands,
而使用 shc_bandshift 时,你应该提供要平移的第一个带,即 shc_bandshift_firstband = num_valence_bands+1。
默认值为 false。
- shc_bandshift_firstband
要平移的第一个导带的索引。
这意味着如果 shc_bandshift 为真,所有索引 i ≥ shc_bandshift_firstband 的能带都将被平移 shc_bandshift_energyshift。
单位是
如果 shc_bandshift 为真,必须提供此标志。
- shc_bandshift_energyshift
导带的能量平移量。
单位为
如果 shc_bandshift 为真,必须提供此参数。
DOS 模块参数
kpath 模块参数
kslice 模块参数
gyrotropic 模块参数
BoltzWann 模块参数
通用能带插值(geninterp)模块参数
Wannier90自带的实用程序
解压Wannier90的压缩包后,这些程序位于路径:
wannier90-3.1.0/utility
**kmesh.pl**
能够生成所需的 kpoints 块内指定网格的 k 点列表
要运行次程序,只需在对应路径运行
/kmesh.pl nx ny nz
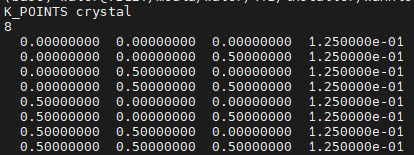
会在输出中生成一个包含 k 点的列表,格式为 Quantum Espresso 格式,其中(除了标题行外)每行的前三个列是 k 坐标,第四列是每个 k 点的权重
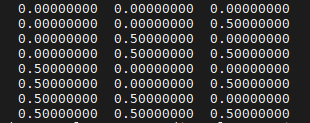
例如运行 ./kmesh.pl 2 2 2 后在当前终端输出:
如果要生成适合 seedname.win 格式 的k 点的列表(即不含权重),运行命令改为
./kmesh.pl nx ny nz wannier
输出的内容可以直接复制到 seedname.win 文件中 kpoints block内
例如运行 ./kmesh.pl 2 2 2 wannier 后在当前终端输出:
*k_mapper.py*
要使用 Yambo 代码进行 GW 计算,需要在不可约布里渊区的网格上执行 nscf 计算。
可能需要比用于插值能带结构更精细的网格来使 GW 计算收敛。k_mapper.py 工具有助于找到完整网格中 k 向量的索引,以便将其插值到用于 Yambo GW 计算的简化网格中。
path/k_mapper.py nx ny nz QE_nscf_output
QE_nscf_output 是传递给 Yambo 的 QE 的 nscf 输出文件的路径
gw2wannier90.py
能够按照能量对 wannier90 的输入数据(例如重叠矩阵和能量本征值)进行排序。
path/gw2wannier90.py seedname options
可用的 options 有
mmn, amn, spn, unk, uhu, uiu,
spn_formatted, unk_formatted, uhu_formatted, uiu_formatted,
支持二进制(非格式化 Fortran)文件,但由于其依赖于编译器,因此不推荐使用。更安全的选择是使用(较大)的格式化文件,选项包括:spn_formatted、uiu_formatted、uhu_formatted、unk_formatted。
write_formatted
默认情况下,输出格式与输入格式相同。要从非格式化输入生成格式化文件,请使用选项:write_formatted
如果没有指定任何选项,则认为所有文件(mmn、amn、spn、UNK、uHu、uIu)均有效。
w90spn2spn.x
第一性原理代码与 wannier90(例如 pw2wannier90.x)之间的接口可以生成一个 .spn 文件,该文件由 postw90 用于计算一些与自旋相关的量。
.spn 文件可以采用依赖于机器的格式或独立于机器的格式进行编写,这取决于 pw2wannier90.x 的输入参数 spn_formatted(默认值为 false,这意味着.spn 文件采用依赖于机器的格式)。如果在一个机器上以依赖于机器的格式生成了.spn 文件,然后想要在另一台机器上(或使用不同编译器编译的 postw90)继续进行计算,那么首先需要在第一台机器上将该文件转换为独立于机器的格式。此时就需要使用 w90spn2spn.x 可执行文件
w90chk2chk.x
如果想在一台机器上运行 wannier90,然后在另一台机器上(或者使用不同编译器编译的 postw90)继续计算,那么首先需要在第一台机器上将 .chk文件转换为与机器无关的“格式化”格式,然后再在第二台机器上转换回来。
为此,请使用 w90chk2chk.x 可执行文件。
PL_assessment
通过在单个单胞上使用密集的 k 点网格进行计算,来评估周期性系统中主层(在Landauer-Buttiker 量子电导计算的背景下)的长度。
assess the length of a principal layer (in the context of a Landauer-Buttiker quantum conductance calculation) of a periodic system using a calculation on a single unit cell with a dense k-point mesh.
./PL_assess.x nk1 nk2 nk3 num_wann
nk1 是 x 方向上的 k 点数量,nk2 是 y 方向上的 k 点数量,nk3 是 z 方向上的 k 点数量,
num_ wann 是系统的 Wannier 函数数量。
!!当前的版本仅允许在与传输方向垂直的方向上有一个 k 点。
然后需要根据提示输入seedname
将返回一个输出文件 seedname-pl.dat,其中包含四列。
第一列:Unit cell number, R
第二列:平均 ’on-site’ 矩阵元,表示主单胞中的 MLWFs 与距其 R 格矢远离的单胞之间的关系。
第三列:第二列的数量的标准差
第四列:第二列中的最大绝对值
w90vdw
提供了一种基于通过最大局域化分波函数(MLWFs)进行密度分解的思想来计算范德华能van der Waals energies 的方法
w90pov
在 utility/w90pov 中提供了一个用于创建 Pov 文件(以便使用 PovRay 工具渲染 Wannier 函数)
的实用程序。
基础知识
周期性体系通过求解周期布洛赫态 来获得电子结构。
除了通过求解布洛赫态,还有一种方法可以求解周期性结构的电子结构,那就是求解Wannier函数(WF)的空间局域化函数(spatially localised functions)。在Wannier90中采用Marzari 和 Vanderbilt(MV) 提出的方法计算最大局域化 Wannier 函数(maximally-localised Wannier functions,MLWF)
对于以晶格点 为中心的Wannier函数
,可以写为布洛赫态的组合:
其中 为晶胞体积,
为酉矩阵,其混合了每个k值下的布洛赫态,不同的
选择会导致波函数具有不同的空间局域性。整个积分在布里渊区(BZ)内进行。
定义WF的展宽 为
总展宽能够分解为一个与gauge(规范)无关的项 加上一个依赖于gauge选择即
的项
,
可以进一步分解为WF 基的对角元
与非对角元
最大局域化 Wannier 函数(MLWF)的构建采用MV方法,通过最小化与规范变换相关的波函数展宽量(即通过优化幺正变换矩阵
来实现)。
Wannier90 在计算时,需要从初始电子结构计算scf中获取两个关键信息:
-
布洛赫态
晶胞周期部分之间的重叠
其中矢量b(用于连接给定k点与其邻近k点)由Wannier90根据文献[1]所述方法确定。 -
作为初始猜测,布洛赫态
上的投影
、
和
都是较小的 N × N 矩阵,与用于获取初始布洛赫态的基组设置无关。