2025深圳杯(东三省)数学建模竞赛D题完整分析论文(共36页)(含模型、可运行代码、数据结果)
2025深圳杯数学建模竞赛D题完整分析论文
目录
摘 要
一、问题重述
二、问题分析
三、模型假设
四、符号定义
五、问题一模型的建立与求解
5.1 问题一模型的建立
5.1.1 问题建模背景
5.1.2 特征工程设计
5.1.3 分类模型结构与数学表达
5.2 问题一模型的求解
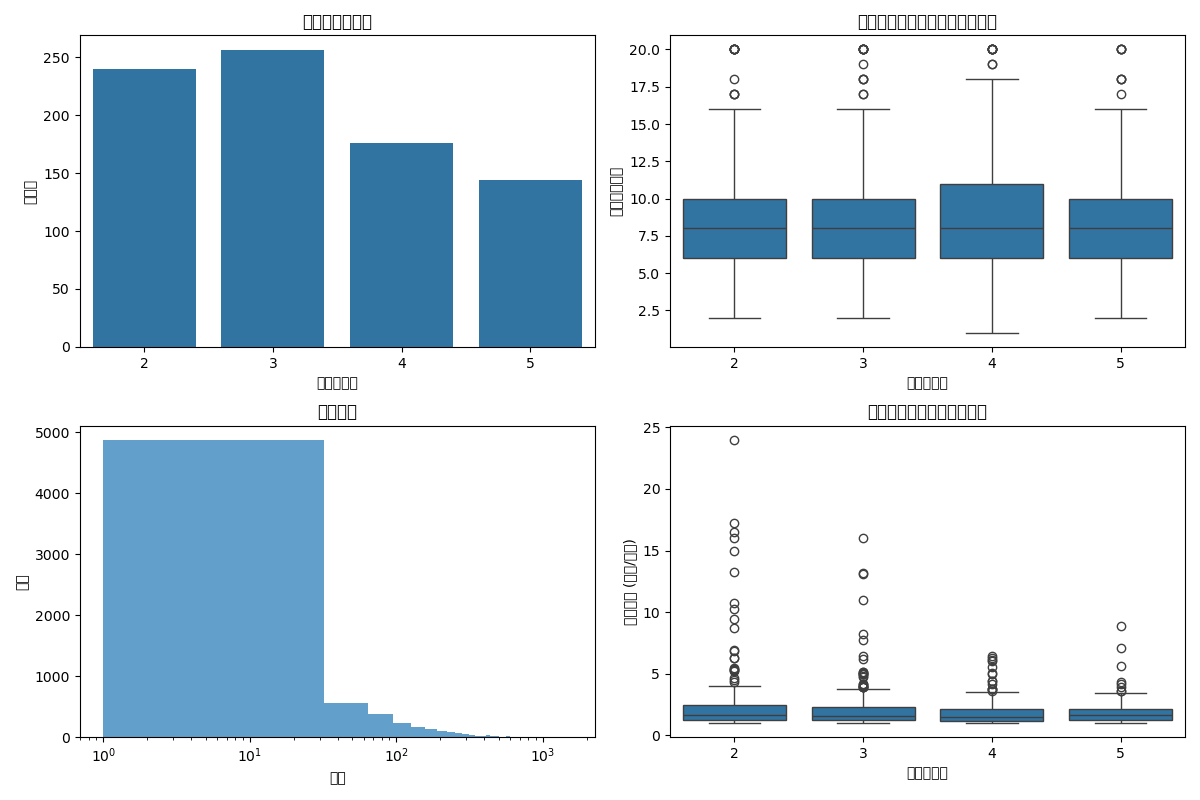
5.2.1描述分析
5.2.2模型总体性能对比
5.2.3 各模型在不同贡献者数量上的表现
六、问题二模型的建立与求解
6.1 问题二模型的建立
6.1.1特征工程设计
6.1.2模型结构与分类器构建
6.1.3模型评估指标定义
6.2 问题二模型的求解
6.2.1数据分布概览
6.2.2模型性能比较
6.2.3重要特征分析
七、问题三模型的建立与求解
7.1 问题三模型的建立
7.1.1贡献者兼容性筛选
7.1.2混合比例优化估计
7.1.3组合匹配评分函数
7.1.4定量评估指标:
7.2 问题三模型的求解
7.2.1数据与参考基因型概览
7.2.2测试样本选择
7.2.3 性能评分
八、问题四模型的建立与求解
8.1 问题四模型的建立
8.2 问题四模型的求解
九、 模型推广
2025深圳杯数学建模D题助攻资料下载链接
https://pan.baidu.com/s/1uWPKN_Jr8SlZV1dtraI66w?pwd=ny2c提取码: ny2c
https://pan.baidu.com/s/1uWPKN_Jr8SlZV1dtraI66w?pwd=ny2c
基于特征工程与机器学习的STR图谱识别研究
摘 要
STR(短串联重复序列)分析是法医学中进行个人身份鉴定的核心技术,其应用广泛,特别是在多名犯罪嫌疑人或受害者的生物样本混合的情境下。混合STR图谱往往包含多个等位基因和复杂的峰型特征,这对传统的身份鉴定方法提出了巨大挑战。如何从这些复杂的峰型图谱中准确判别贡献者人数、估计各贡献者的DNA比例,以及还原各自的基因型,是提高法医鉴定准确性和效率的关键问题。因此,这一问题在法医学领域具有重要的理论意义和广泛的司法应用价值。
针对问题一,我们首先提出了一个多类分类问题,旨在识别混合样本中贡献者的数量。为此,我们提取了包括等位基因总数、各等位基因峰高的统计特征(如均值、标准差、极值、峰高排序比值等)以及片段长度分布等结构化特征,应用于不同的机器学习模型进行分类学习。实验结果表明,GBDT(梯度提升树)模型在识别2人混合样本时表现最佳,但在3人及以上混合样本中,由于等位基因重叠较为严重,识别难度大大增加。整体而言,采用多模型集成的分类策略能够有效捕捉不同贡献者人数对STR图谱特征的影响,其中GBDT模型在准确性和效率方面最为突出。
针对问题二,即已知贡献者人数情况下的DNA比例估计,我们通过特征工程方法,计算每个位点的有效等位基因数、峰高的均值、标准差、变异系数等统计特征,以表征各贡献者的比例结构。在已知贡献者人数(2至5人)的前提下,我们将不同的比例情况视作离散类别,并分别训练针对不同人数的比例分类器。实验结果表明,2人混合样本的比例识别准确率较高,但随着贡献者人数增加,识别准确率显著下降,尤其在3人及以上样本中,比例趋于均衡时,等位基因的重叠增加了分类难度,现有方法在这些复杂场景下仍有很大的改进空间。
针对问题三,我们提出了一种基于兼容性评分与匹配度量的组合搜索方法,用于推断混合样本中每位贡献者的基因型。该方法首先计算每位候选个体基因型与混合样本中观测到的等位基因的兼容性评分(即重合等位基因的比例),接着针对选定的贡献者组合,优化估计其混合比例,使得预测峰高与观测峰高之间的差异最小。最终,通过设计综合匹配评分函数,将等位基因解释率和峰高分布相关性相结合,对候选组合进行排序。得分最高的个体组合即为最可能的贡献者,其在每个基因座上的等位基因即为推断的基因型输出。该方法以兼顾等位基因匹配和峰高拟合为核心,能够有效提高推断精度,并通过准确率、召回率等指标评估结果的可靠性。
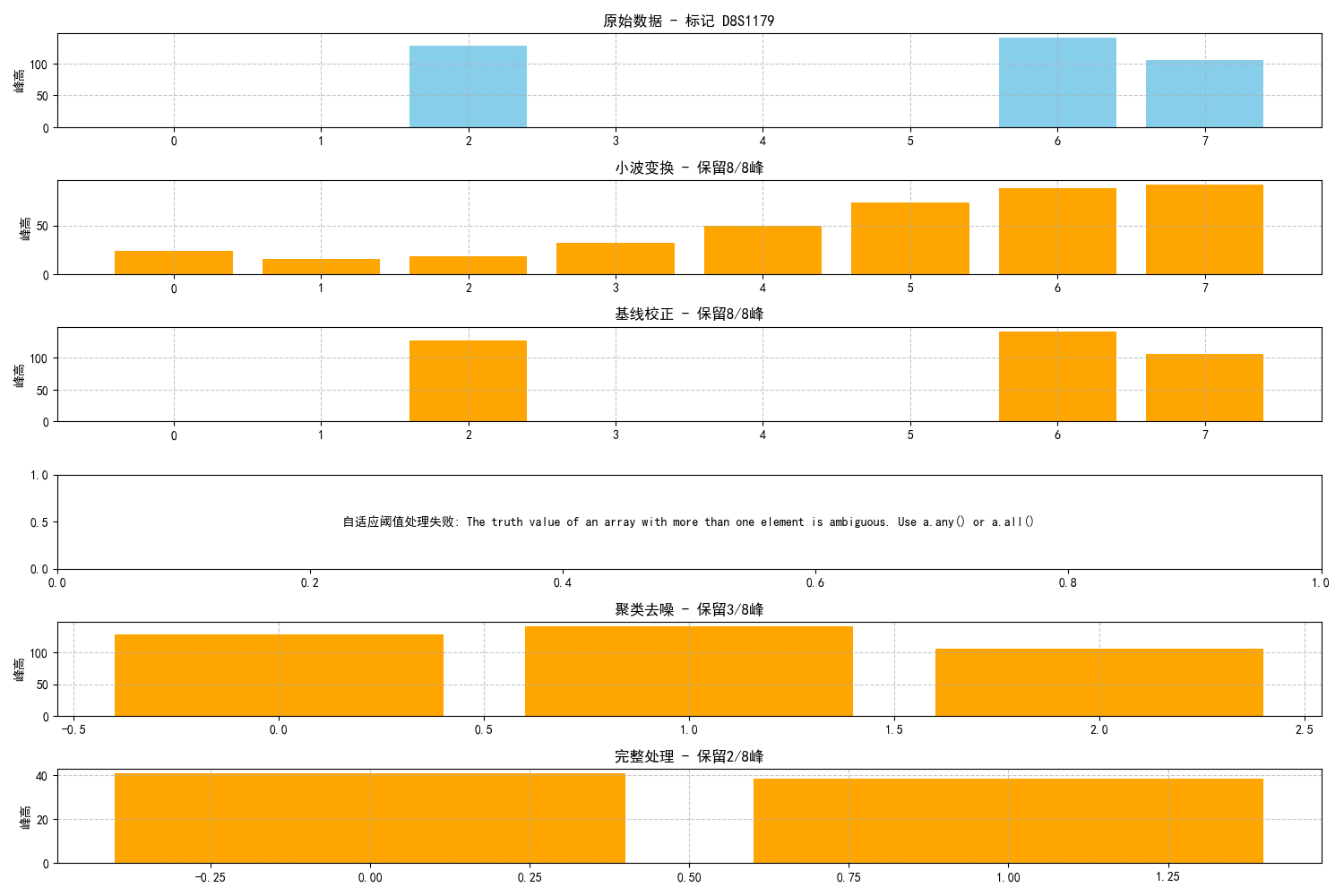
针对问题四,我们提出了一种多阶段改进型去噪流程,结合信号处理与匹配算法,以有效减少混合样本中噪声的干扰,提高分析的准确性。该流程首先采用小波阈值去噪方法对原始峰高信号进行处理,以抑制高频噪声并尽可能保留真实的峰信号;接着,识别并筛除可能的拖尾峰(PCR扩增伪峰);然后,通过自适应评分机制筛选出可靠的峰值,并采用改进的匹配得分函数对贡献者进行识别。实验结果表明,该去噪方法显著提高了样本的贡献者识别准确率,在大多数样本中准确率可达到0.8至1.0,即使在某些极端样本中,该方法也优于仅做预处理或传统去噪的方案。总体而言,该流程有效抑制了噪声干扰,提高了混合样本解析的鲁棒性和准确度。
关键词: STR;GBDT;峰高均值;鲁棒性;小波阈值去噪
2025深圳杯数学建模D题助攻资料下载链接
https://pan.baidu.com/s/1uWPKN_Jr8SlZV1dtraI66w?pwd=ny2c提取码: ny2c
提取码: ny2c百度网盘 请输入提取码
一、问题重述
1.1 问题的背景与意义
DNA短串联重复序列(STR)分析是法医学中用于个人身份鉴定的核心技术,广泛应用于刑事案件侦破、灾难身份识别、亲子鉴定等领域。STR技术通过分析个体基因组中特定位点的重复次数,利用其高度的多态性和遗传稳定性,实现对不同个体的区分与识别。然而,法医物证中往往存在多人混合的DNA样本,例如性侵、斗殴等案件中,多个涉案者留下的生物物证会导致DNA成分的混合。与单一来源的样本不同,混合DNA样本中的STR图谱呈现出复杂的峰型特征,可能在同一基因座上检测到多个等位基因,且每个等位基因的峰高反映了不同贡献者的DNA比例。如何从这些复杂的图谱信息中准确判断贡献者人数、估算各贡献者的DNA比例以及还原每个贡献者的基因型,成为提升法医鉴定准确性和效率的关键问题。
解决这些问题对提高法医物证分析的准确性具有重大意义,不仅能有效协助司法机关精准锁定犯罪嫌疑人和受害者,还能为案件定性提供科学依据。同时,这一研究方向的深入推进,也能够促进法医自动化、智能化和标准化的发展,为数据挖掘、统计推断和机器学习方法在法医学领域的应用提供了丰富的数据支持和实践场景。
1.2 问题的提出
本研究针对法医领域中STR混合数据的分析与应用,主要聚焦于以下几个方面:
1. 混合样本中贡献者人数的判别
混合DNA样本中,贡献者人数通常未知。通过对给定的STR图谱数据进行分析,需要设计有效的算法或统计方法,自动识别并判定混合样本中包含多少名不同的贡献者。准确识别贡献者人数对于后续数据分解和个人身份识别的精度至关重要。
2. 混合样本中各贡献者DNA比例的估计
多人混合DNA样本中,每位贡献者的DNA比例通常不同。利用STR图谱中各个位点的等位基因峰高数值,如何精确估算每个贡献者的DNA比例,是突破传统人工经验估算、提升数据解释客观性的关键。合理的比例估算能够有效减少误差,并提高分析的准确性。
3. 结合已知数据库信息的身份匹配与验证
在某些案件中,调查人员能够获得嫌疑人或受害人的STR基因型信息。如何结合这些已知信息与混合样本数据,通过模型判断相关个体是否为混合样本的贡献者,并优化混合比例的估算方案,是实际案件侦查中的重要课题。身份匹配与验证不仅能帮助确定嫌疑人身份,还能提供有力的证据支持。
4. 自动化、可推广的数据分析模型建立
随着法医学中高通量、大规模混合STR样本数据的出现,构建一套自动化、鲁棒性强的全流程数据分析模型显得尤为重要。该模型应能够从贡献者人数的推断、比例的估算到身份匹配,提供一个端到端的解决方案。通过提升法医工作效率、实现标准化和智能化,该模型不仅能提升分析精度,还能推动相关数据挖掘、统计推断和人工智能方法在法医学领域的广泛应用,推动产学研的融合与发展。
2025深圳杯数学建模D题助攻资料下载链接
https://pan.baidu.com/s/1uWPKN_Jr8SlZV1dtraI66w?pwd=ny2c提取码: ny2c
提取码: ny2c百度网盘 请输入提取码
二、问题分析
2.1 问题一分析
混合STR样本中的贡献者人数判别任务被建模为多类分类问题。通过特征工程,从STR图谱中提取与贡献者人数相关的统计特征,如等位基因数量、非OL等位基因数量、峰高的均值、标准差、变异系数、前几高峰占比及片段长度等信息。接着,我们采用多种机器学习模型,包括梯度提升树(GBDT)、随机森林、支持向量机(SVM)和多层感知器(MLP)等,通过对这些特征的学习,完成贡献者人数的识别。GBDT模型因其对数据的处理能力尤其在面对小规模不平衡样本时的表现较好,因此成为了最优选择。然而,在处理3人及以上的混合样本时,由于等位基因重叠较为严重,识别的准确性下降。因此,特征提取和模型优化是解决此类问题的关键。
2.2 问题二分析
估计已知贡献者人数条件下的DNA比例是另一个重要任务。我们将比例估计视作离散的比例类别分类问题,对于2至5人混合样本,分别设计适合的比例类别,如2人组合可能包括“1:4、1:1、1:9”等。通过提取STR图谱中的等位基因数量、峰高统计、片段长度等特征,我们训练了多个模型来进行比例估算。在实验中,发现GBDT适用于2人比例估计,MLP适用于3人,而随机森林在4人和5人混合样本中表现较为出色。模型的选择基于其在处理不同维度和非线性关系中的优势,GBDT在小规模不平衡样本中表现强劲,MLP则擅长捕捉复杂的模式,随机森林具有较强的鲁棒性。随着贡献者人数的增加,比例的准确性下降,尤其是在比例接近的情况下,等位基因重叠增加了分类的难度。
2.3 问题三分析
在已知候选个体基因型的数据库条件下,推断混合样本中各贡献者的基因型是一个关键任务。我们提出了一种基于兼容性筛选和匹配评分的逐步求解策略。首先,计算每个候选个体在核心基因座上的兼容性得分,即个体基因型与混合样本中观测等位基因的重合度(完全重合则兼容,部分重合则扣分)。对兼容性较高的个体组合进行枚举或搜索,并通过加权最小二乘法估算其混合比例,最小化预测峰高与观测峰高的差异。最终,我们设计了综合匹配评分函数,将等位基因解释率与峰高相关性结合,对组合进行排序,得分最高的组合即为最优解。该方法能够有效筛选出最可能的贡献者,并恢复他们在每个位点上的等位基因组合。然而,如何精确估算混合比例,尤其是多贡献者的复杂组合,仍然是一个挑战。
2.4 问题四分析
混合STR图谱中的噪声干扰对数据分析的准确性有显著影响,尤其是在复杂样本中。我们设计了一种多模块的降噪框架来解决这一问题。首先,利用小波阈值去噪方法对原始峰高信号进行处理,通过小波分解将信号分解到不同频带,并对高频系数进行软阈值处理,从而抑制高频噪声,保留低频信号。接着,针对PCR扩增伪峰(拖尾峰),我们设计了基于峰高比值和片段长度差的识别规则,通过这些规则识别并筛除可能的伪峰。然后,通过自适应峰值筛选机制对候选峰进行质量评分,筛除低质量峰。最后,经过去噪处理后的信号被输入到改进的匹配得分函数中,用于贡献者识别。该方法有效提高了贡献者识别和基因型还原的准确性,尤其在极端或复杂样本中表现出较强的鲁棒性。去噪技术的优化仍是提升准确性的关键因素。
详细情况可以看一下视频或点击链接
2025深圳杯数学建模D题助攻资料下载链接
https://pan.baidu.com/s/1uWPKN_Jr8SlZV1dtraI66w?pwd=ny2c提取码: ny2c
提取码: ny2c百度网盘 请输入提取码