LIANA | part1 intro部分
本文是一篇综述(review),核心工作是比较了cell-cell communication inference的16个resources和7个methods,文章的核心就是这个,对全文的理解都要基于这两份比较。
我对resources是什么具体还不是很理解,于是我在文章中进行了这样的挖掘:
1、文章intro部分提到,这篇文章讨论的CCC是这样的:
CCC commonly refers to interactions between secreted ligands and plasma membrane receptors. This picture can be broadened to include secreted enzymes, extracellular matrix proteins, transporters, and interactions that require the physical contact between cells, such as cell-cell adhesion proteins and gap junctions. For simplicity, we refer to all of these events involving protein-protein interactions as CCC.
A number of computational tools and resources have emerged that can be further classified as those that predict CCC interactions alone, and those that additionally estimate intracellular activities related to CCC.Here, we focus on the former (Table 1).
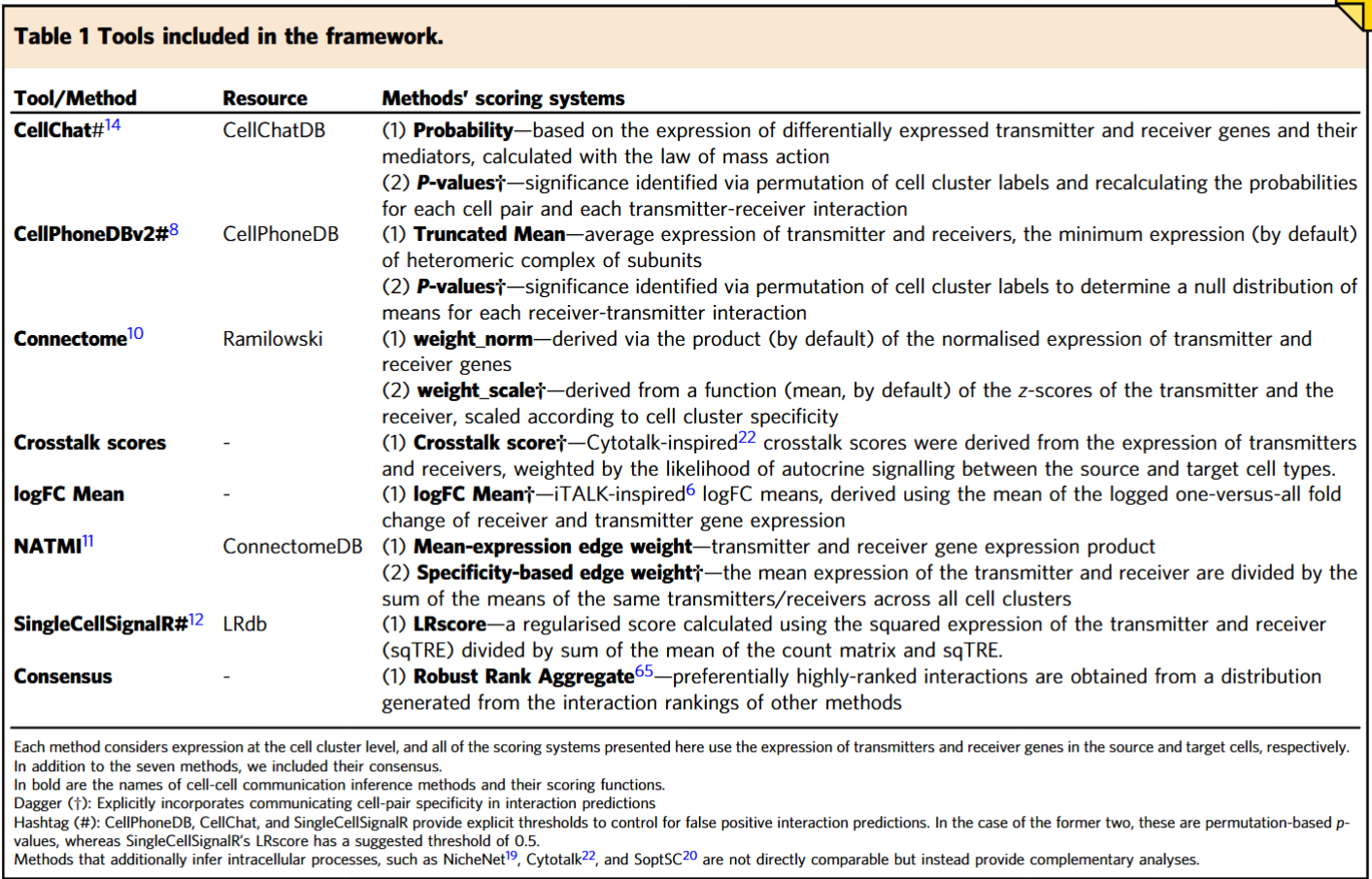
看到这里其实就很明确了,经典的CCC events在本质上是protein-protein interactions,我们把信号的发出方的protein叫做ligands,把信号的接收方的protein叫做receptors。
These CCC tools typically use gene expression information obtained by scRNA-Seq. In general, single cells are clustered by their gene expression profile and cell type identities are assigned to the clusters based on known gene markers. Then, CCC tools can predict intercellular crosstalk between any pair of clusters, one cluster being the source and the other the target of a CCC event. CCC events are thus typically represented as a one-to-one interaction between a transmitter and receiver protein, accordingly expressed by the source and target cell clusters. The information about which transmitter binds to which receiver is extracted from diverse sources of prior knowledge.
这里进一步往前推,把CCC的分析从“protein的分子层面”上升到了“cluster的细胞层面”,也就是我们看到的很多scRNA-seq数据出来之后的分析中,都会做的Clustering和Differential Expression Gene analysis,它们的核心目的是给某一堆细胞贴一个标签,这个标签一般是细胞大类的名称,部分是亚型的名称,具体怎么给细胞做Annotation也有非常多的学问。做完annotation之后,就会得到说,和protein分为ligand-receptor类似,cluster也分为sourse cluster与target cluster,这种one-to-one的interaction,我们的视角一下就提升到了cluster层面,关注的是哪一群细胞和另外哪一群细胞之间在发生信息传递。
这里也很关键地提到了prior knowledge,但我目前仍然不清楚prior knowledge到底是蛋白质层面的知识还是cell层面的知识。于是看到了这一段:
The available prior knowledge resources, largely composed of ligand-receptor, extracellular matrix, and adhesion interactions, are typically distinct but often show partial overlap. Some of these resources also provide additional details for the interactions such as information about subcellular localisation, classification into signalling pathways and categories (Supplementary Table 1). Notably, some resources (Supplementary Table 1), and consequently their corresponding methods, focus on protein complexes as the functional units of CCC, which are crucial for the coordination of signalling as different subunit combinations may induce distinct responses. Despite the fact that CCC inference is constrained by the prior knowledge used, yet the impact of resource choice is largely unexplored, with the exception of a descriptive comparison of 4 resources with one method. Thus, it remains unclear how the choice of resource and method affects the results and thereby the biological interpretation of the scRNA-seq data.
这里很明显就表达清楚了prior knowledge的具体含义,比如ligand-receptor pair配受体对、extracellular matrix细胞外基质、adhesion interactions黏附作用,部分resources还提供了subcellular localisation亚细胞定位、classification into signalling pathways and categories信号通路归属及功能划分(这里涉及大量signalling pathway的知识,于是把信号通路的归属于功能写并一起)。还有一些方法对protein做了细分,也就是CCC的unit不只是两个蛋白质而已,而是两份protein complexes,需要考虑到蛋白质亚基的问题。
到这里就清晰很多了,我们所谓的先验知识prior knowledge,核心是ligand-receptor的配对的信息,以及帮助确定cell所处微环境、cell所处空间位置、cell所在信号通路等的信息。
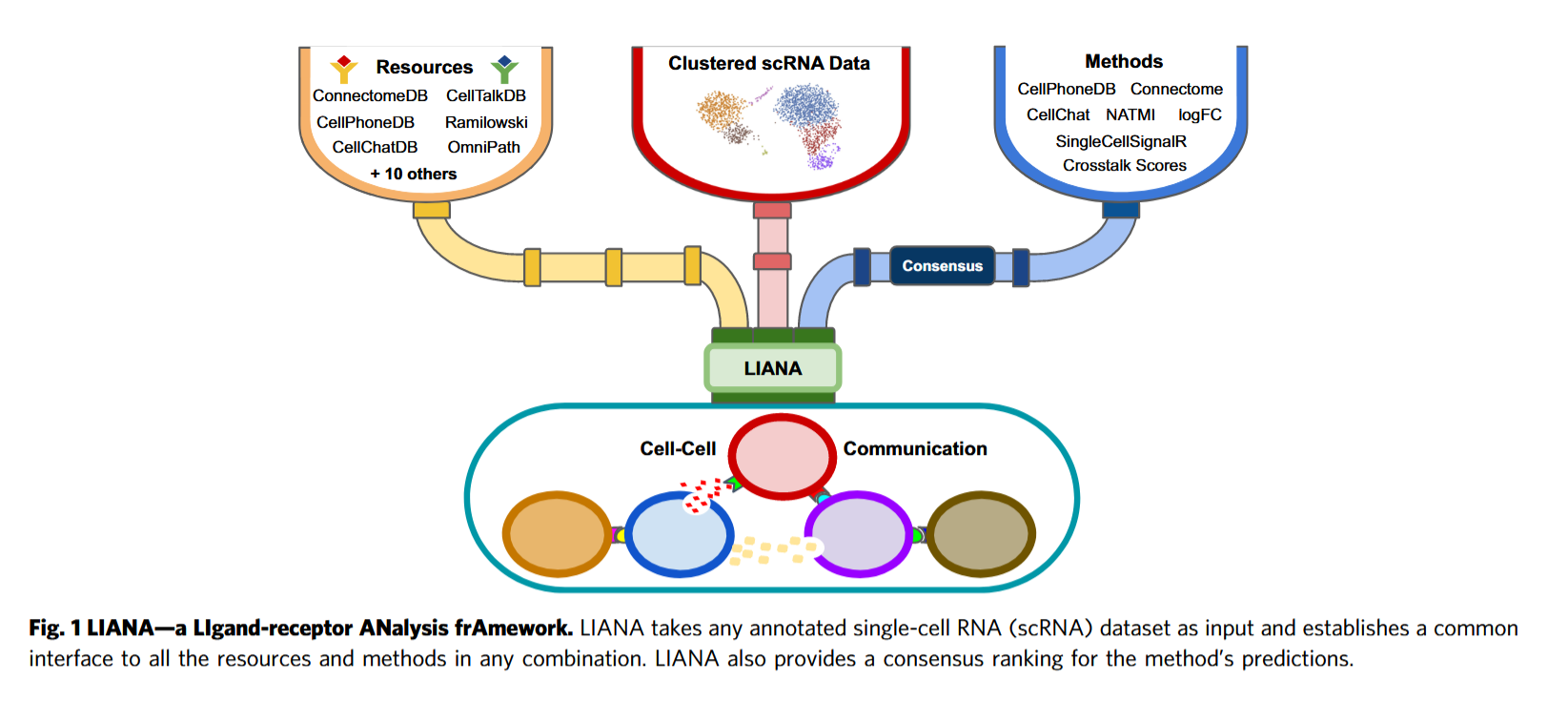
可以看到正是LRP先验知识、scRNA-seq data、analysis methods三管齐下,共同撑起了cell-cell communication的分析。
2、但是我对resources具体长什么样还是不太清楚,于是我去查了我感兴趣的两个DataBase
CellPhoneDB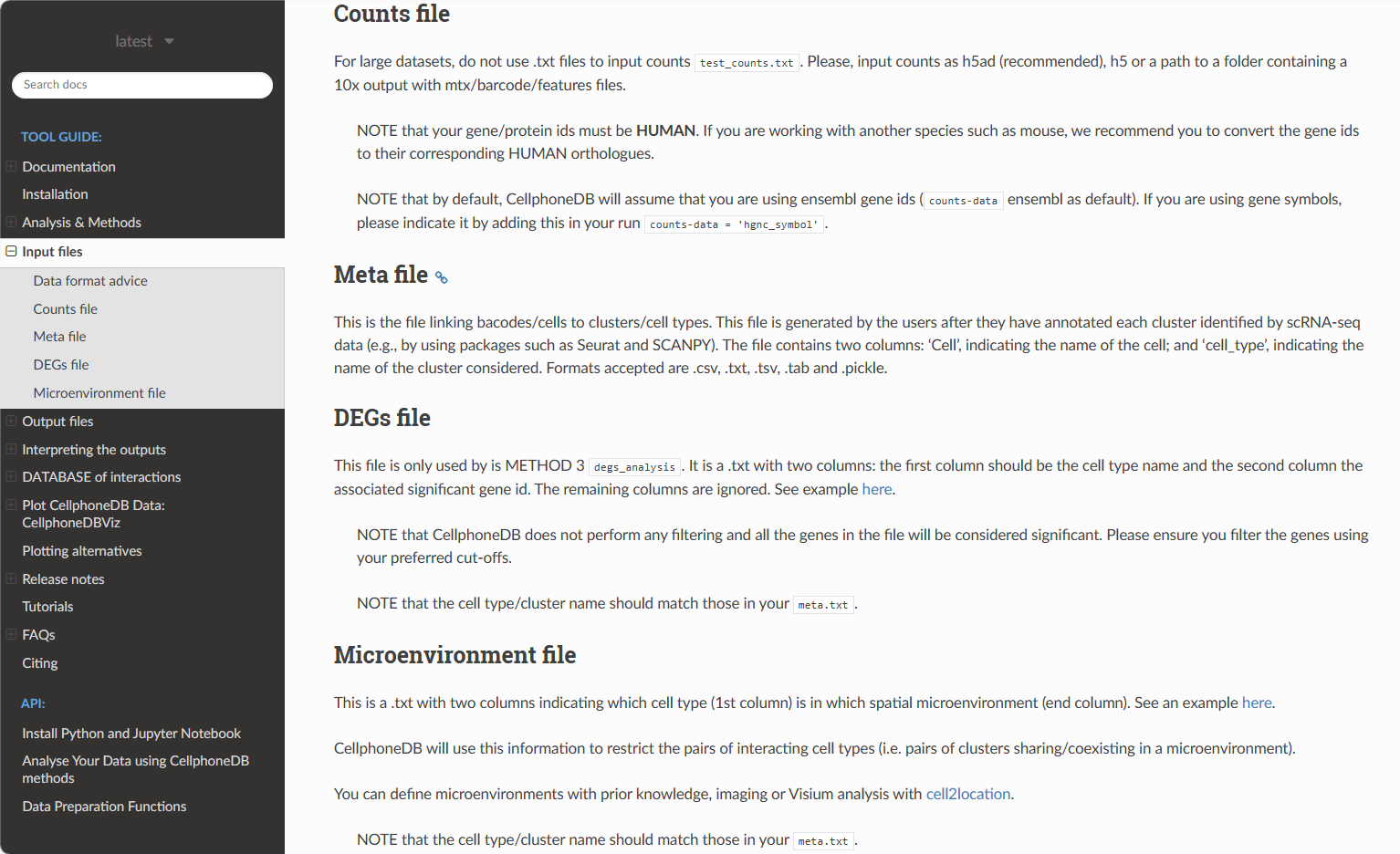
我让AI帮我总结了这四种file的大致内容:
📁 CellphoneDB 输入文件要求解析
1. Counts文件(计数数据文件)
用途:存储大型数据集的基因表达计数数据
格式要求:
- ❌ 不推荐:
.txt文件 - ✅ 推荐:
.h5ad格式(最佳选择) - ✅ 可选:
.h5格式 - ✅ 可选:包含10x输出的文件夹路径(含mtx/barcode/features文件)
重要注意事项:
- 🧬 基因ID必须是人类基因ID
- 🐭 如使用小鼠等其他物种数据,需转换为对应的人类同源基因
- 🏷️ 默认基因ID格式:Ensembl gene IDs
- 🔤 如使用基因符号,需在运行时指定:
counts-data = 'hgnc_symbol'
2. Meta文件(元数据文件)
用途:链接细胞条码与细胞类型/聚类的对应关系
文件结构:
- 📋 包含两列:
Cell:细胞名称/条码cell_type:聚类/细胞类型名称
支持格式:.csv, .txt, .tsv, .tab, .pickle
生成方式:用户在使用Seurat、SCANPY等工具完成细胞注释后生成
3. DEGs文件(差异表达基因文件)
用途:仅用于方法3 degs_analysis
文件格式:
- 📝
.txt格式 - 🔢 至少两列:
- 第1列:细胞类型名称
- 第2列:相关的显著基因ID
- 其余列被忽略
重要提醒:
- ⚠️ CellphoneDB不进行任何过滤
- 📊 文件中所有基因都被视为显著基因
- ✂️ 用户需预先使用首选阈值过滤基因
- 🏷️ 细胞类型名称必须与
meta.txt中的一致
4. Microenvironment文件(微环境文件)
用途:定义细胞类型的空间微环境关系
文件格式:
- 📝
.txt格式 - 🔢 两列结构:
- 第1列:细胞类型
- 第2列:所属空间微环境
功能:
- 🔗 限制相互作用的细胞类型对
- 🎯 仅考虑共存于同一微环境的细胞聚类对
微环境定义方法:
- 🧠 先验知识
- 🔬 成像分析
- 📍 使用cell2location进行Visium分析
注意事项:细胞类型名称必须与meta.txt中的匹配
💡 关键要点总结
- 数据格式优先级:h5ad > h5 > 10x文件夹 > txt(不推荐)
- 物种兼容性:必须转换为人类基因ID
- 文件命名一致性:所有文件中的细胞类型名称必须保持一致
- 数据预处理:用户需自行完成基因过滤和细胞注释
由于我并没有跑过很多流程,所以我对这些file的理解不是很到位。但这些似乎并不是所谓LRP的先验知识。于是我进入了文章原文CellPhoneDB v5: inferring cell–cell communication from single-cell multiomics data | Nature Protocols
在这里找到了v5版本的Databasehttps://github.com/ventolab/CellphoneDB-data
里面的数据是这个样子的(这里是一个Ensembl文件):
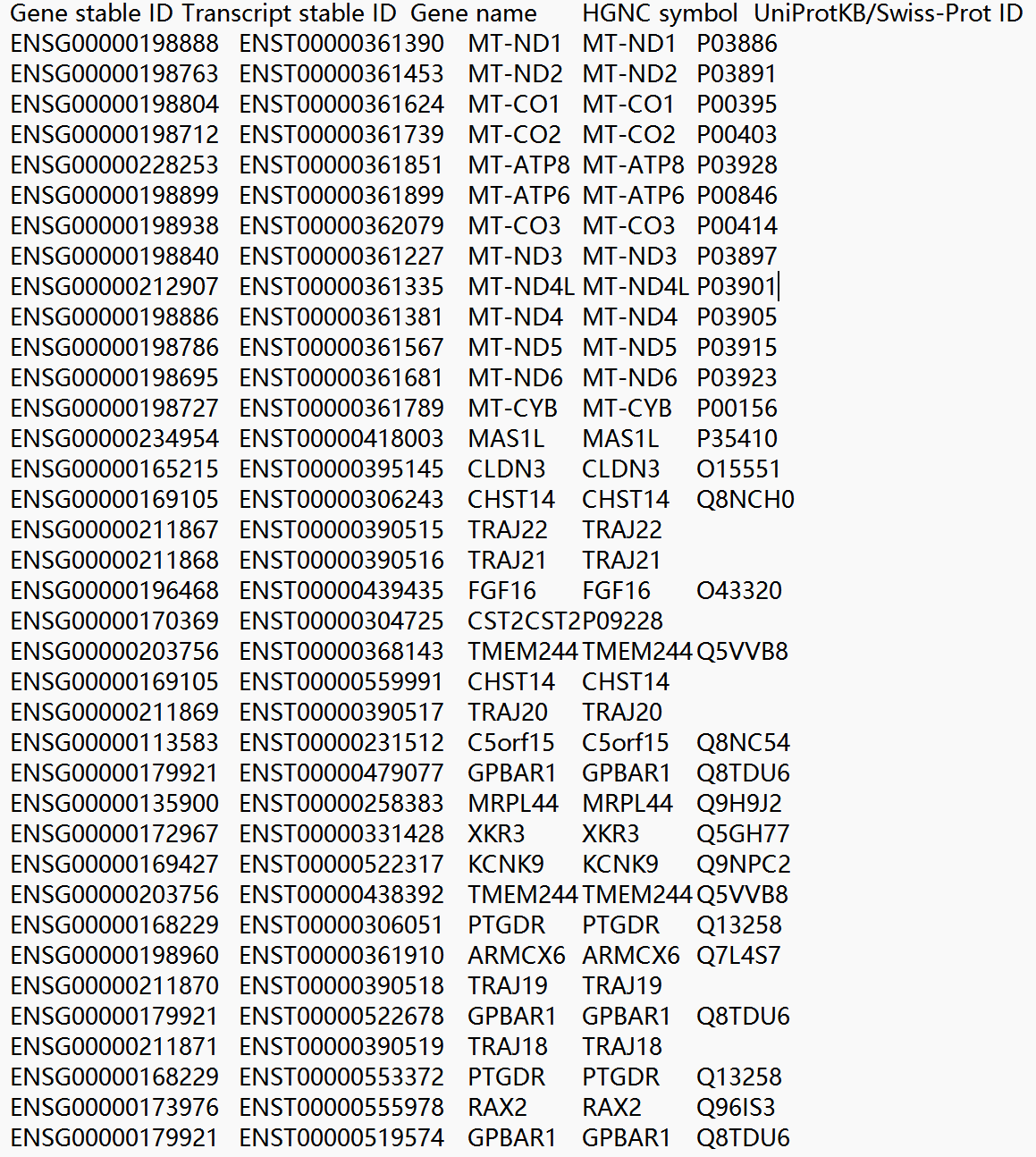
📂 CellphoneDB v5.0.0 数据结构解析
1. 总体文件夹架构
cellphonedb-data-5.0.0/
├── data/
│ ├── sources/ # 原始数据源文件
│ ├── complex_input # 蛋白质复合体输入文件
│ ├── gene_input # 基因输入文件
│ ├── interaction_input # 相互作用输入文件
│ ├── protein_input # 蛋白质输入文件
│ └── transcription_factor_input # 转录因子输入文件
2. Sources文件夹内容分析
📁 sources/ 文件夹包含三个关键文件:
ensembl- Ensembl基因注释数据uniprot.tab- UniProt蛋白质数据库信息uniprot_synonyms.tsv- UniProt同义词映射表
3. Ensembl文件详细解析
从您展示的ensembl文件数据表格,我可以看到包含以下字段:
🧬 字段结构分析:
| 字段名 | 示例数据 | 含义 |
|---|---|---|
| Gene stable ID | ENSG00000198888 | Ensembl基因稳定标识符 |
| Transcript stable ID | ENST00000361390 | Ensembl转录本稳定标识符 |
| Gene name | MT-ND1 | 基因名称 |
| HGNC symbol | MT-ND1 | HUGO基因命名委员会符号 |
| UniProtKB/Swiss-Prot ID | P03886 | UniProt蛋白质数据库ID |
🔍 数据内容特征:
1. 线粒体基因丰富
- 大量以
MT-开头的线粒体基因 - 如:MT-ND1, MT-ND2, MT-CO1, MT-ATP8等
- 这些是细胞呼吸链复合体的关键组分
2. 基因类型多样
- 线粒体基因: MT-ND1, MT-CO1, MT-ATP6等
- 膜蛋白基因: CLDN3, TMEM244等
- 信号分子: FGF16, TRAJ22等
- 受体相关: PTGDR, KCNK9等
3. ID映射完整性
- 提供了从Ensembl ID到HGNC符号的完整映射
- 包含UniProt蛋白质ID,便于跨数据库整合
4. 数据用途和重要性
🎯 在CellphoneDB中的作用:
-
基因ID标准化
- 将不同命名系统统一为HGNC标准
- 支持Ensembl ID和基因符号的互转
-
蛋白质功能注释
- 通过UniProt ID获取蛋白质功能信息
- 支持复合体组装的准确建模
-
物种转换支持
- 为其他物种数据转换为人类同源基因提供基础
- 确保跨物种分析的准确性
📊 数据统计信息:
- 文件大小:22.7 MB
- 包含字符数:5,219,651个
- 涵盖了人类基因组的大部分已注释基因
5. 实际应用建议
💡 使用该数据时的关键点:
-
基因ID转换
# 示例:查找基因对应关系 ensembl_to_hgnc = {'ENSG00000198888': 'MT-ND1','ENSG00000165215': 'CLDN3',# ... 更多映射 } -
数据质量检查
- 验证您的单细胞数据中的基因ID格式
- 确保与CellphoneDB期望的ID类型匹配
-
物种转换准备
- 如果使用非人类数据,需要预先转换为对应的人类同源基因
这个ensembl文件是CellphoneDB数据库的核心组件之一,为准确的细胞间通讯分析提供了标准化的基因注释基础。
然后我接着打开了uniprot.tab文件与uniprot_sunonyms.tsv文件(居然是在adobe illustrator打开的?):

📋 UniProt数据文件详细解析
1. uniprot.tab 文件结构分析
| 字段名 | 示例数据 | 含义说明 |
|---|---|---|
| Entry | A0A584, Q9BXU3 | UniProt唯一标识符 |
| Entry name | TVBK2_HUMAN, TX13A_HUMAN | 蛋白质条目标准名称 |
| Status | reviewed | 数据审查状态(reviewed=已审查,高质量) |
| Protein names | T cell receptor beta variable 11-2 | 蛋白质全名描述 |
| Gene names | TRBV11-2, TEX13A | 对应的基因名称 |
| Organism | Homo sapiens (Human) | 物种信息(均为人类) |
| Length | 115, 409, 903 | 蛋白质氨基酸序列长度 |
🧬 数据内容特征:
1. 受体和信号分子丰富
- T细胞受体: TRBV11-2 (T cell receptor beta variable)
- 激酶类: TXK (Tyrosine-protein kinase)
- 转录因子: TXNL1 (Thioredoxin-like protein)
2. 多样化的蛋白质功能
- 细胞膜受体: 多种受体蛋白
- 酶类: 激酶、磷酸酶等
- 信号传导分子: 硫氧还蛋白相关蛋白
3. 高质量数据标准
- 所有条目均为"reviewed"状态
- 提供完整的功能注释
- 包含精确的序列长度信息
2. uniprot_synonyms.tsv 文件结构分析
🔍 字段详细解读:
| 字段名 | 示例数据 | 含义说明 |
|---|---|---|
| Entry | A0A024QZ08, A0A024QZ26 | UniProt ID |
| Entry Name (primary) | A0A024QZ08_HUMAN | 主要条目名称 |
| Protein names | Intraflagellar transport 20 homolog | 蛋白质功能描述 |
| Gene Names (synonym) | (Chlamydomonas), isoform CRA_c | 基因名称同义词 |
| Gene Names | IFT20, HDAC6, TBX2 | 标准基因名称 |
📊 同义词映射功能:
1. 基因名称标准化
IFT20 ← Intraflagellar transport 20 homolog
HDAC6 ← Histone deacetylase 6
EPHA2 ← receptor protein-tyrosine kinase
2. 异构体信息
isoform CRA_c,isoform CRA_a: 不同的蛋白质异构体- 帮助区分同一基因的不同转录本产物
3. 功能分类覆盖
- 转录调控: HDAC6 (组蛋白去乙酰化酶)
- 信号转导: EPHA2 (受体酪氨酸激酶)
- 细胞结构: IFT20 (鞭毛内转运蛋白)
3. 两文件的协同作用
🔗 数据整合功能:
-
ID映射链
Ensembl ID → HGNC Symbol → UniProt ID → 蛋白质功能 -
多层次注释体系
- 基因层面: Ensembl + HGNC
- 蛋白质层面: UniProt + 功能描述
- 同义词层面: 历史名称 + 别名映射
-
质量控制机制
- 仅包含reviewed状态的蛋白质
- 提供完整的交叉引用
- 支持多种命名系统互转
4. 在CellphoneDB中的重要性
🎯 核心应用价值:
1. 受体-配体识别
# 示例:受体蛋白识别
receptor_proteins = {'EPHA2': 'receptor protein-tyrosine kinase','NR4A1': 'Nuclear receptor subfamily 4 group A member 1'
}
2. 蛋白质复合体建模
- 通过UniProt ID获取亚基信息
- 构建异源多聚体复合物
- 验证相互作用的生物学合理性
3. 功能注释增强
- 为预测的相互作用提供功能背景
- 支持生物学意义的结果解释
- 减少假阳性预测
5. 数据使用建议
💡 最佳实践:
1. 数据预处理
# 建议的数据处理流程
def process_uniprot_data():# 1. 加载主要UniProt数据uniprot_main = pd.read_csv('uniprot.tab', sep='\t')# 2. 加载同义词映射uniprot_synonyms = pd.read_csv('uniprot_synonyms.tsv', sep='\t')# 3. 创建统一的ID映射字典id_mapping = create_unified_mapping(uniprot_main, uniprot_synonyms)return id_mapping
2. 质量验证
- 优先使用"reviewed"状态的蛋白质
- 验证基因名称的一致性
- 检查蛋白质长度的合理性
3. 功能富集分析
- 利用蛋白质功能描述进行通路分析
- 识别特定细胞类型的关键受体-配体对
- 构建功能模块网络
📈 数据统计概览:
- 数据质量: 全部为reviewed状态
- 物种专一性: 100%人类蛋白质
- 功能覆盖: 涵盖细胞通讯相关的主要蛋白质家族
- 更新频率: 与UniProt数据库同步
这两个文件构成了CellphoneDB蛋白质注释体系的核心,为准确的细胞间通讯分析提供了可靠的分子基础。