【Cell Systems】SpotGF空间转录组去噪算法文献分享
目录
题目
摘要
介绍
结果
结果1:SRT数据中存在的噪声会阻碍下游分析的开展。
结果2:SRT数据的去噪方案SpotGF
SpotGF 在实际数据中的工作原理与性能验证
SpotGF评分准确反映基因扩散程度的验证
结果3:SpotGF 能有效过滤SRT数据中的扩散基因
SpotGF 提升聚类精度的原因解析
SpotGF 在模拟噪声与真实场景中的去噪优势
结果4:SpotGF 提高了SRT数据中上调基因鉴定的准确性
结果5:SpotGF 有助于提升聚类分析效果与细胞类型推断准确性
结果6:SpotGF 提高了在人类结直肠癌中识别肿瘤细胞的准确性
SpotGF 提升了肿瘤相关上调基因的比例与特异性
讨论
空间分辨转录组学(SRT)数据中的噪声问题与 SpotGF 的解决方案
SpotGF:通过过滤“无效基因”实现 SRT 数据去噪
SpotGF 作为识别空间变异基因(SVGs)的强大工具
SpotGF:提升 SRT 数据质量,助力精准生物学解读
放在前面
文章链接:SpotGF: Denoising spatially resolved transcriptomics data using an optimal transport-based gene filtering algorithm: Cell Systems
工具github链接:GitHub - illuminate6060/SpotGF: SpotGF: Denoising Spatially Resolved Transcriptomics Data Using an Optimal Transport-Based Gene Filtering Algorithm
其他相关链接:GitHub - illuminate6060/SpotGF_data_form_change
这篇发表到cell systems(IF:9.3,Q1)上,并入选为了封面文章。与大家一起学习~


题目
SpotGF:基于最优传输的基因过滤算法用于空间转录组学数据去噪研究
摘要
空间分辨转录组学(SRT)将基因表达谱与细胞在其天然状态下的物理位置相结合,但由于冷冻切片过程中细胞受损以及染色和mRNA释放试剂的暴露,容易受到不可预测的空间噪声影响。为解决这一问题,我们开发了 SpotGF——一种基于最优传输的基因过滤算法,用于对空间分辨转录组学数据进行去噪。SpotGF通过数值化量化扩散模式,区分广泛表达基因与聚集表达基因,并将前者作为噪声进行过滤。与传统去噪方法不同,SpotGF保留原始测序数据,从而避免了因插补可能产生的假阳性结果。此外,SpotGF在聚类分析、潜在标记基因识别及细胞类型注释方面均展现出卓越的性能。总体而言,SpotGF有望成为空间分辨转录组学数据下游分析中一个关键的预处理步骤。
介绍
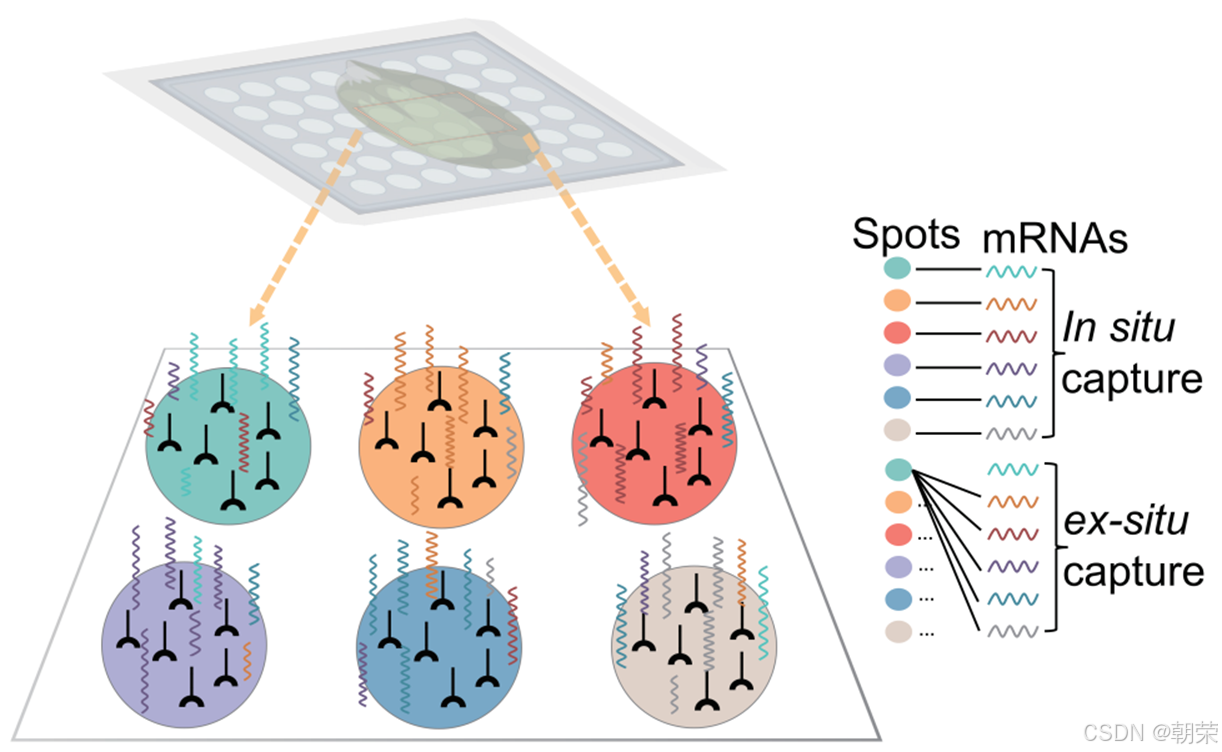
空间分辨转录组学(SRT)技术将高通量基因测序与组织学技术相结合,在提供基因表达数据的同时,还能结合细胞壁荧光染料Calcofluor White(CW)或细胞核染料4,6-二脒基-2-苯基吲哚二盐酸盐(DAPI)等染色图像,反映细胞在空间中的分布信息。SRT数据通过包含空间位置信息,有助于分析细胞类型,推动细胞功能发现、细胞互作研究等多种分析工作。理想情况下,每个位于特定位置的测序单元(在不同技术中称为“珠子”或“位点”)应仅捕获原位细胞释放的转录本。然而,由于实验环境中液相条件下的随机扩散,SRT也可能捕获非原位的转录本。这种扩散会在SRT数据中引入复杂的噪声,其复杂程度甚至超过单细胞RNA测序(scRNA-seq)中常见的“dropout”现象。
导致SRT数据噪声的主要因素有三:
首先,在组织冷冻切片后,冷冻切片会被固定在空间转录组芯片的表面以进行透化处理。这些芯片旨在捕获细胞释放的信使RNA(mRNA),但若细胞透化不完全,则会阻碍芯片上位于细胞下方的位点捕获mRNA,导致这些位点上方的mRNA发生随机漂移;相反,如果细胞透化过度,则会导致大量mRNA被释放,这些mRNA可能被邻近的位点捕获。此外,由于细胞透化是在液相环境中进行的,受热运动和分子无规则运动的影响,mRNA的漂移是一种随机事件。
其次,在RNA测序的反转录和扩增过程中,也可能发生转录本的扩散。RNA与反转录酶形成的复合物可能会破坏部分RNA或导致其泄漏。不当的结合或反应条件可能在cDNA合成过程中通过引物诱导RNA扩散。
最后,在SRT数据处理过程中,若采用不恰当的算法、参数或质量控制标准,也会增加假阳性噪声。此外,SpotClean曾深入讨论过组织和背景之间的转录本交换问题,而Sprod则在10X Visium卵巢癌数据集中验证了复杂的空间噪声存在。
针对SRT数据中的噪声,研究者已尝试多种解决方法。最初,研究人员采用了为解决scRNA-seq中“dropout”问题而开发的表达插补策略,例如Magic、scImpute和SAVER。然而,这些方法对于SRT数据中具有空间相关性的噪声效果有限。随后,研究者开发了专门针对SRT数据空间噪声的定制算法。例如,Sprod利用匹配位置的位置信息和相应的成像数据来对SRT数据进行去噪;SpotClean则通过概率模型降低噪声,并通过转录本交换调整唯一分子标识符(UMI)计数。这些方法试图通过统计手段模拟SRT数据中的转录本扩散模式,并对原始UMI计数进行修改。然而,受实验操作影响的随机且无方向性的转录本扩散,使得对这些模式的准确建模面临挑战。上述基于统计的算法主要聚焦于数据修改和插补,因而可能在SRT数据中引入假阳性表达,掩盖那些低表达的原位基因。此外,实验噪声和假阳性数据还会干扰下游的聚类分析,特别是在Louvain和Leiden等社区检测算法中表现尤为明显。
因此,我们提出将SRT数据中的基因分为两类:一类是具有明显空间聚集特征的基因,视为有效信号(即“有效基因”);另一类是具有高扩散水平的基因,视为噪声(即“无效基因”)。为了减轻转录本扩散所带来噪声的影响,我们通过排除具有高扩散水平的“无效基因”来增强“有效基因”的信号。我们证明,在开展任何下游分析之前,进行此类去噪处理具有至关重要的意义。
结果
结果1:SRT数据中存在的噪声会阻碍下游分析的开展。
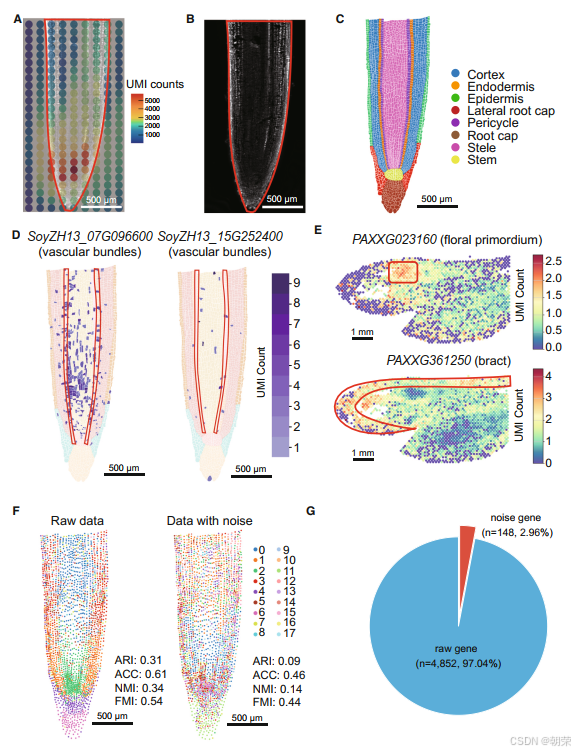
由于转录本扩散噪声在SRT数据中普遍存在,我们重点在两种主流的空间分辨转录组学技术——Stereo-seq 和 10X Visium 上对该现象进行了研究。在此,我们验证了Stereo-seq大豆根系数据中UMI计数的扩散现象。在使用200格(100微米方格)的数据时,我们观察到明显的UMI计数延伸到了右侧组织区域之外(图1A和图1B)。随后,我们手动标注了八种主要细胞类型,生成了基于细胞分割结果的细胞-格数据(图1C)。两个预期仅在维管束细胞中特异性表达的标记基因——SoyZH13_07G096600 和 SoyZH13_15G252400,在原始数据中却显示出在整个根系组织中广泛表达(图1D)。
在10X Visium兰花花序数据的三张切片中,我们也观察到了标记基因空间表达模式的漂移现象。在其中一张切片中,预期仅在原基组织中特异性表达的标记基因 PAXXG023160 和 PAXXG009220,展现出了更为广泛的表达(图1E和图S1A)。同样地,与苞片组织相关的标记基因 PAXXG361250 和 PAXXG024270,不仅出现在苞片组织内,还扩散到了苞片组织的边界之外(图1E和图S1A)。这种现象在其他两组兰花数据中也一致出现(图S1B)。类似地,在10X Visium人脑前额叶皮质SRT数据(LIBD_151507)中,我们也发现了两个标记基因存在非原位表达的现象(图S1C)。这些结果表明,SRT数据中普遍存在广泛的转录本扩散,导致基因丧失其原有的空间特异性,从而无法对聚类分析或细胞类型注释产生积极作用。
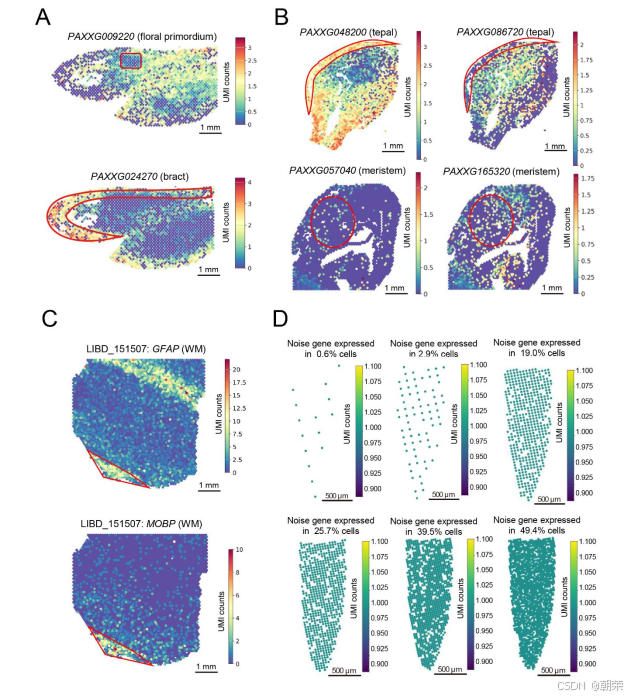
接下来,我们评估了“无效基因”对下游分析的影响。我们使用Scanpy[17]将原始的大豆根系数据聚类为18个群集,得到的评估指标包括:调整兰德指数(Adjusted Rand Index, ARI)为0.31、准确率(Accuracy, ACC)为0.61、归一化互信息(Normalized Mutual Information, NMI)为0.34,以及Fowlkes-Mallows指数(Fowlkes-Mallows Index, FMI)为0.54(图1F和表S1)。为了评估噪声的影响,我们在数据中人为引入了5,000个均匀分布于整个组织内的模拟噪声基因(图S1D和表S2)。结果显示,含有噪声的数据在进行聚类后,所有四项评估指标均出现了下降(图1F),这表明噪声基因会显著削弱聚类分析的效果。进一步地,我们从含噪声的数据中筛选出5,000个高变异基因(Highly Variable Genes, HVGs),发现其中仍有2.96%的模拟噪声基因未被过滤掉(图1G和图S2A-C),这说明常用的高变异基因筛选方法对于这类扩散基因并不有效。因此,在进行下游生物信息学分析之前,对SRT数据进行有效的去噪处理显得至关重要。
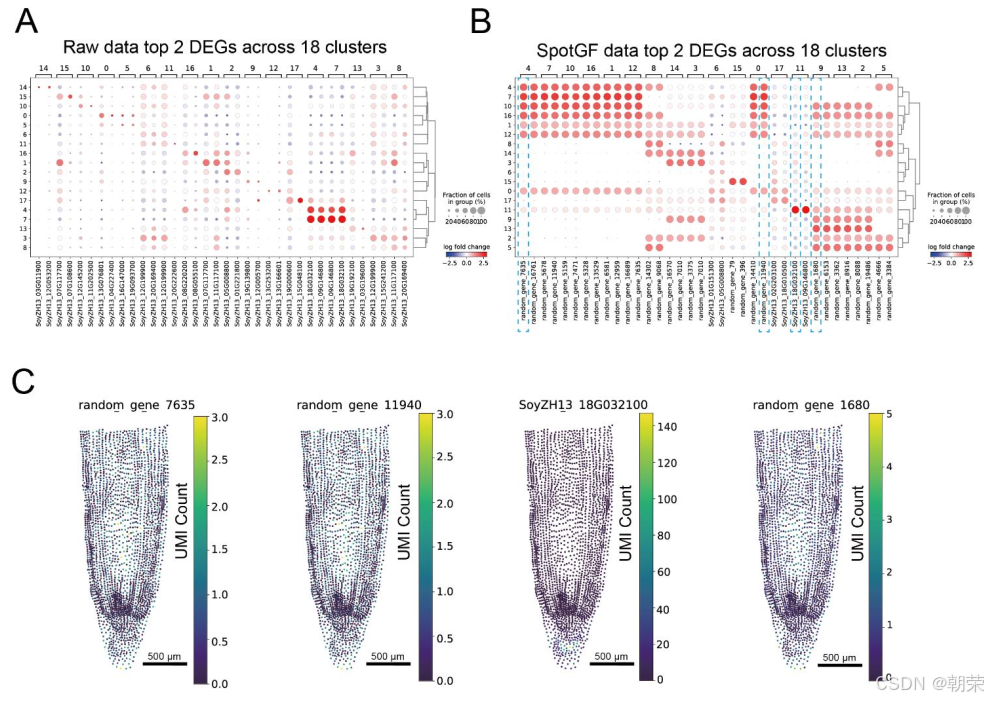
结果2:SRT数据的去噪方案SpotGF
为应对复杂的空间噪声问题,我们开发了 SpotGF 算法。该算法基于最优传输(Optimal Transport, OT)理论,能够定量评估基因的扩散模式(图2A),从而筛选并过滤掉发生扩散的基因,有效降低噪声对数据分析的影响。SpotGF 的核心作用在于识别并过滤掉“无效基因”(图2B)——这类基因通常在组织区域内呈现广泛且均匀的表达,对聚类分析、细胞类型注释以及差异表达基因(DEGs)鉴定等关键任务并无帮助。与依赖数理统计模型的传统去噪算法不同,SpotGF 不会对原始测序数据中的“有效基因”UMI计数进行任何修改,而是专注于剔除那些阻碍下游分析的“无效基因”(图2C)。
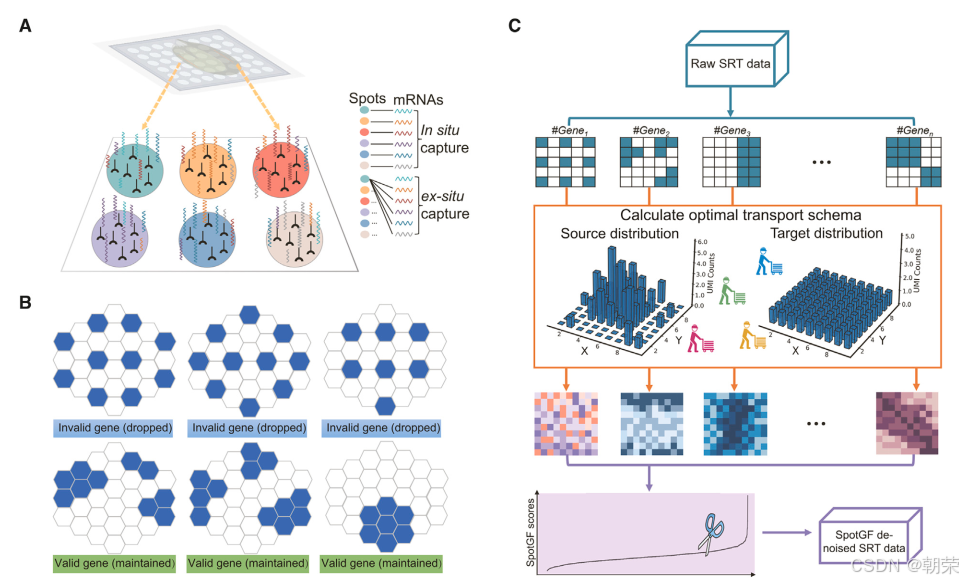
SpotGF 在实际数据中的工作原理与性能验证
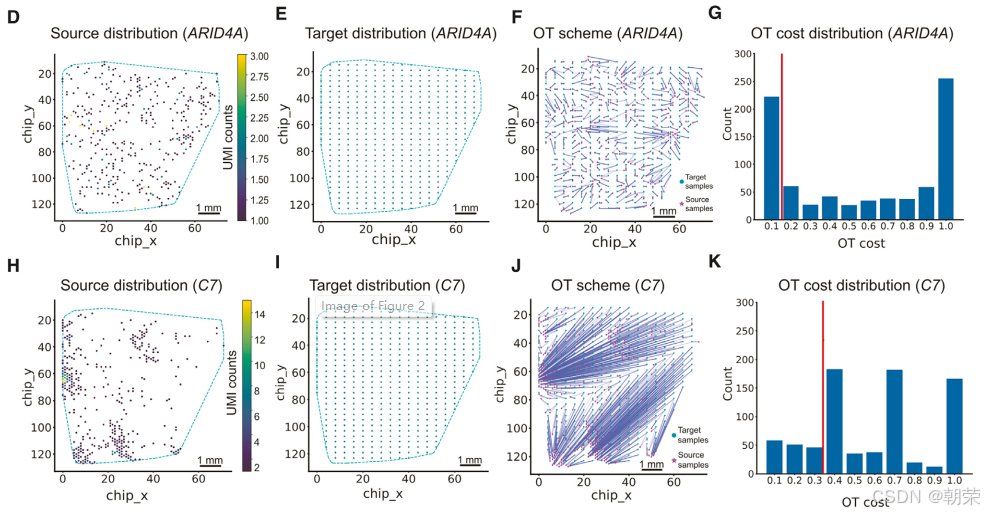
我们以 10X Visium 人结直肠癌数据 为例,选取了两个具有代表性的基因——ARID4A(一种在组织中广泛分布的“无效基因”)和 C7(一种空间聚集分布的“有效基因”)——来评估 SpotGF 的实际性能。
对于基因 ARID4A,SpotGF 首先利用每个位点(spot)的位置信息,构建了一个二维空间分布,称为“源分布”(source distribution,图2D)。接着,采用 alpha-shape 算法对组织轮廓进行估算。基于源分布及其组织轮廓的特征,生成了一个代表最大扩散状态下假设表达情况的“目标分布”(target distribution,图2E)。随后,运用 最优传输(OT)方法计算源分布与目标分布之间的传输成本(transportation cost),进而生成OT方案(图2F)。我们对OT方案中每一步的成本进行了量化(图2G),发现总共经历了800个步骤,累计总成本为 1,473.51。我们将每个基因在最优OT方案中的传输成本作为该基因的 SpotGF评分。
同理,我们对基因 C7 也进行了相同的分析流程:构建源分布(图2H)、生成对应的目标分布(图2I)、计算OT方案(图2J)以及评估每一步的成本(图2K)。结果显示,该基因总共经历了789个步骤,但总成本高达 14,696.70。值得注意的是,基因 ARID4A 的标准化步骤成本大多集中在 0.1~0.2 区间(图2G),而基因 C7 的大部分标准化步骤成本则集中在 0.4 或更高(图2K)。这一差异导致 C7 的总成本远高于 ARID4A,说明 SpotGF评分能够准确反映基因在组织中的表达扩散程度:SpotGF评分越低,表明扩散越严重;评分越高,则意味着基因呈现更强的空间聚集特性。
此外,我们还提供了一个基于梯度变化的自动化算法,用于确定 SpotGF 评分的过滤阈值,并生成经过去噪处理的数据。同时,用户也可以根据自身SRT数据的特性,通过外部参数灵活手动调整该阈值。
SpotGF评分准确反映基因扩散程度的验证
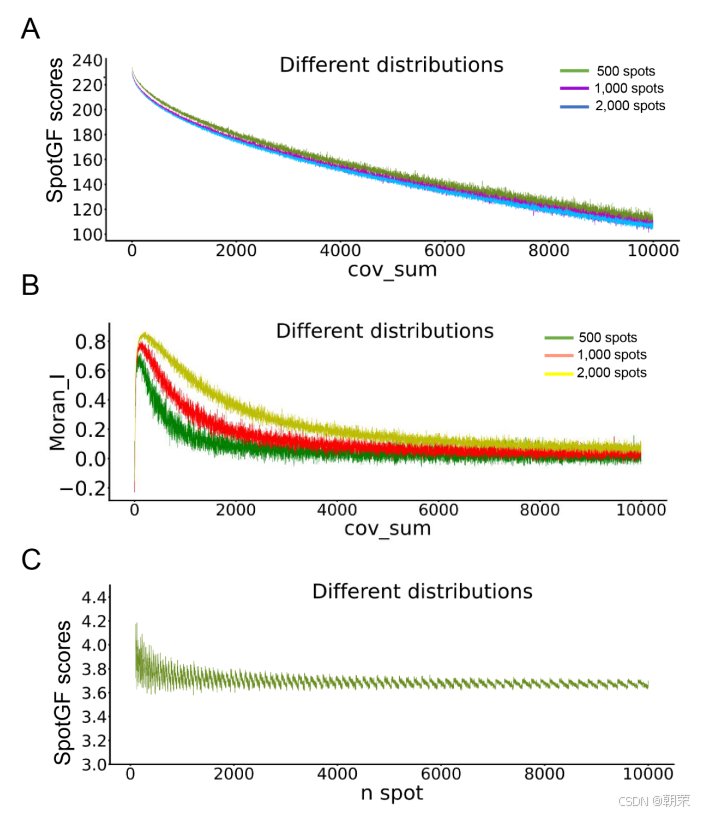
为了验证 SpotGF 评分在衡量基因扩散水平上的准确性,我们生成了三组具有不同扩散程度的二维空间高斯分布(图S3A 和 表S3)。在排除高斯分布中随机干扰因素后,SpotGF 评分与这些空间分布的扩散特征呈现出显著线性相关性(图S4A)。相比之下,常用于评估基因空间自相关性的 Moran's I 指标,在刻画这些分布的扩散特性方面则效果不佳(图S4B)。
为了消除因不同基因位点数量差异带来的影响,我们还生成了 10,000 个模拟基因,这些基因具有相同的扩散特征但位点数量各异。对于位点数量超过10个(通常在质控步骤中被筛除)的空间分布,SpotGF 评分始终稳定在 3.7~3.8 的范围内波动(图S4C 和 表S3)。虽然理论上我们期望评分保持恒定,但由于高斯分布生成过程本身具有一定的随机性,0.1 范围内的波动被认为是可接受的。
结果3:SpotGF 能有效过滤SRT数据中的扩散基因
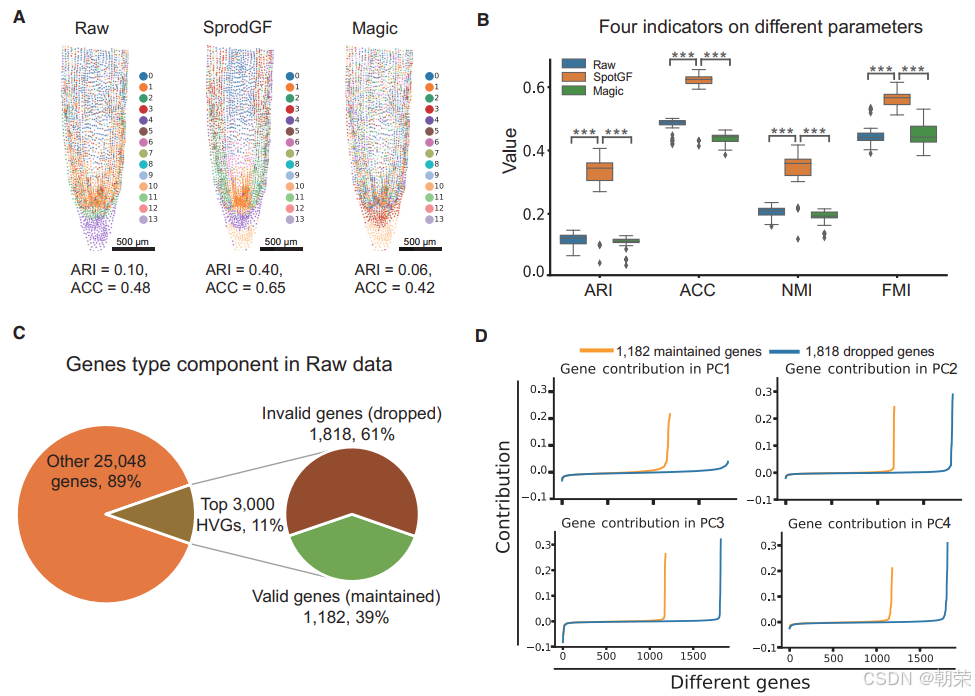
为了评估 SpotGF 在过滤噪声基因方面的有效性,我们将其应用于 Stereo-seq 大豆根系数据。在使用前 3,000 个高变异基因(HVGs)将原始数据、SpotGF 去噪数据以及 Magic 去噪数据分别聚类为 14 个群集后,SpotGF 去噪数据取得了最高的调整兰德指数(ARI = 0.40)和准确率(ACC = 0.65)(图3A、图S5A 及表S1)。此外,在根据每个群集的预测准确率重新分配类别后,SpotGF 去噪数据成功区分出了四种不同的细胞类型(图S5B)。
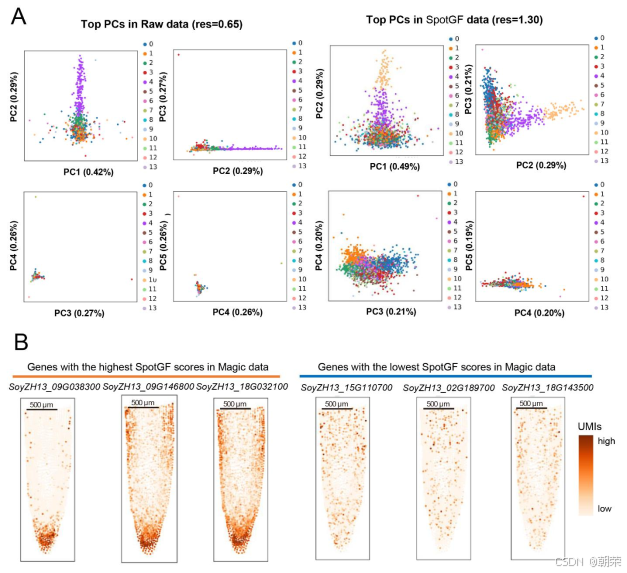
在探索 78 种不同参数组合时,SpotGF 去噪数据在四个评估指标上始终优于其他数据集(图3B、图S5C 及表S4)。为了理解原始数据与 SpotGF 去噪数据之间的差异,我们生成了每个细胞中总 UMI 计数的热图,发现 SpotGF 去噪数据在特征表现上比 Magic 去噪数据更接近原始数据(图S5D 和图S5E)。通过主成分分析(PCA)得到的前五个主成分(PCs)的分布模式显示,SpotGF 去噪数据所衍生的主成分在区分不同细胞类型方面更具效力(图S6A)。这些结果充分证明了 SpotGF 在降低 SRT 数据噪声方面的有效性。
SpotGF 提升聚类精度的原因解析
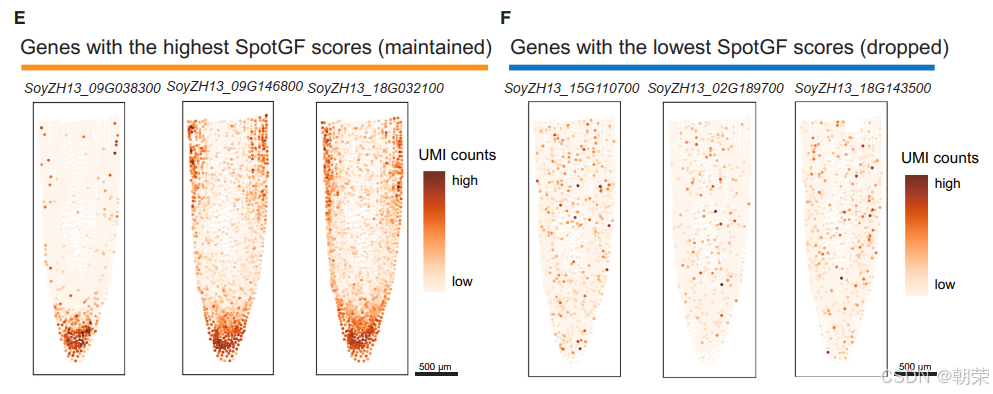
为了解释为何 SpotGF 去噪数据能获得更高的聚类精度,我们分析了原始数据中的前 3,000 个高变异基因(HVGs),发现其中 61%(1,818 个基因)应当被剔除,而 39%(1,182 个基因)则应当保留(图3C)。通过 PCA 进行定量评估后发现,SpotGF 去噪数据中所保留的基因在最优主成分 PC1 中具有更高的贡献率,从而增强了 PC1 区分不同细胞类型的能力;而那些被剔除的基因若被保留,其较高的贡献率反而会削弱 PC2、PC3 和 PC4 在细胞区分上的能力。此外,随着主成分索引的增加,保留基因的贡献率逐渐下降(图3D)。这些发现揭示了噪声对降维分析的负面影响,强调了在降维过程中进行基因过滤的必要性。
此外,我们还展示了 SpotGF 评分最高的前三个基因(具有显著空间聚集特性) 的空间分布情况(图3E),以及 SpotGF 评分最低的前三个基因(呈现全组织范围内的均匀表达) 的分布(图3F)。相比之下,Magic10 方法未能改善基因表达的空间聚集特性(图S6B)。
SpotGF 在模拟噪声与真实场景中的去噪优势
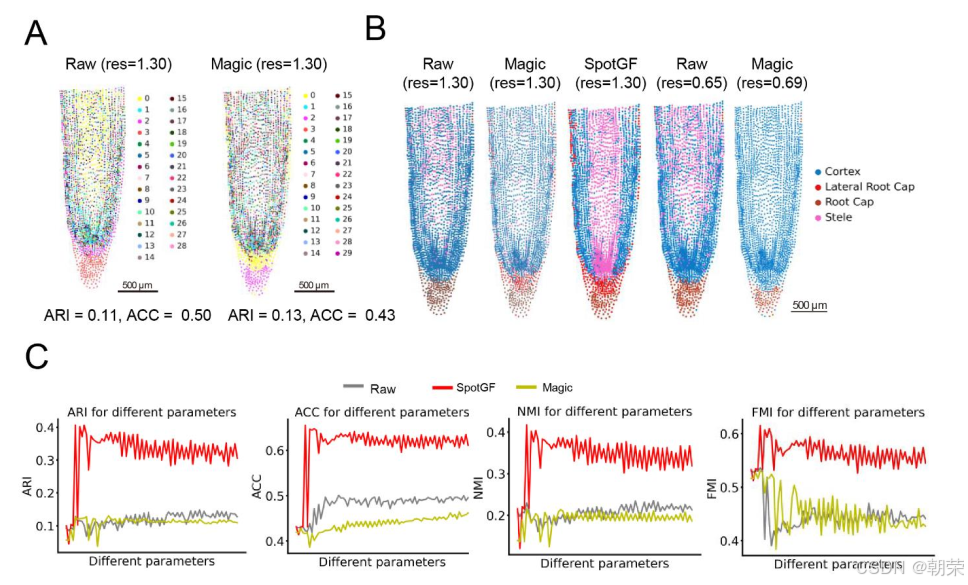
我们还进一步证明,在使用前 5,000 个 HVGs 进行聚类时,SpotGF 去噪数据所得到的聚类结果最接近真实情况,成功识别出五种细胞类型(图S7A 和图S7B)。
此外,我们向大豆根系数据中引入了 5,000 个在全组织范围内均匀表达的模拟噪声基因,并分别对原始含噪声数据及经过 SpotGF、Magic、Sprod、SpotClean 和 STAGATE[25] 等方法去噪后的数据进行了 21 个群集的聚类分析。结果显示,SpotGF 是处理这些噪声基因最优的去噪方法(图S7C 和图S7D)。
结果4:SpotGF 提高了SRT数据中上调基因鉴定的准确性
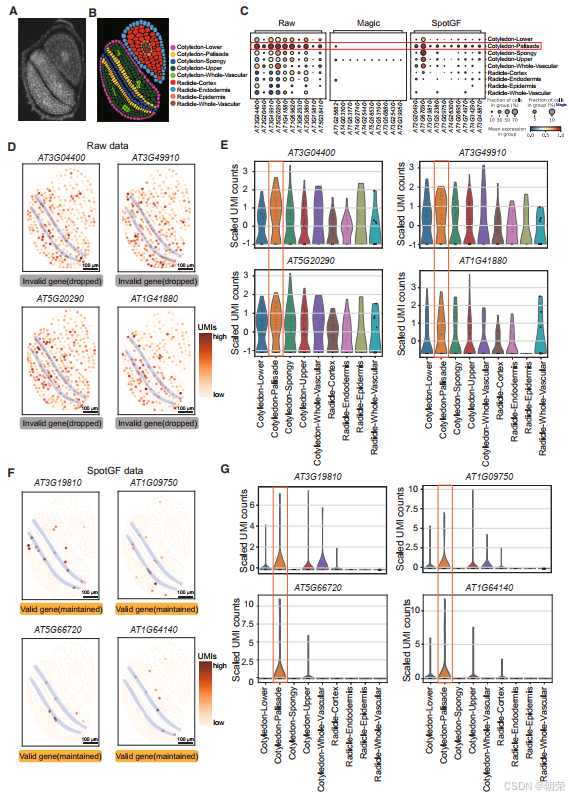
与传统方法通过无监督聚类来识别细胞类型特异性基因不同,SRT 数据允许我们直接根据细胞的形态学特征来识别细胞类型,然后通过差异表达分析找出每种细胞类型的特征基因。首先,我们基于形态学信息,生成了拟南芥胚胎的细胞-位点(cell-bin)数据,并根据 FB 染色图像标注了九种细胞类型,这些标注同时也作为可靠的真实参考标准(图4A 和图4B)。我们以此为基础,验证了 SpotGF 在存在扩散现象的拟南芥 SRT 数据中识别上调基因的有效性。
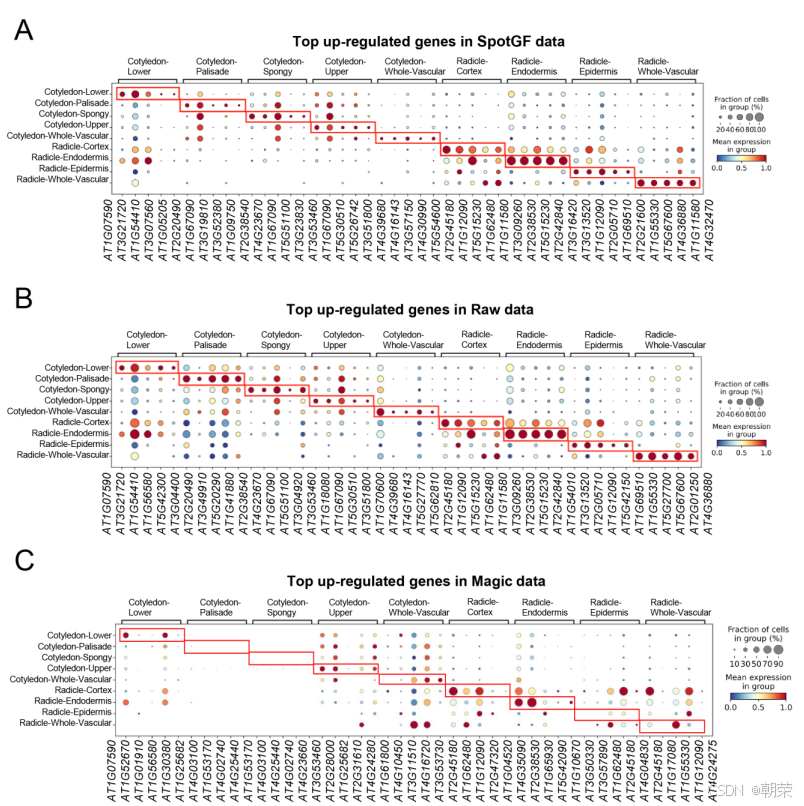
我们使用 Scanpy分别在九种细胞类型中筛选出了上调程度排名前五的基因。结果发现,从 SpotGF 去噪数据中识别出的上调基因,相比原始数据和 Magic 去噪数据,具有更高的细胞类型特异性(图4C 和图S8A-C)。例如,在原始数据中识别出的栅栏细胞(palisade cells)上调基因表现出很高的扩散性(图4D 和图4E),而从 SpotGF 去噪数据中识别出的上调基因则显示出很高的空间特异性,并且在其他八种细胞类型中的表达水平较低(图4F 和图4G)。此外,从 Magic 去噪数据中识别出的上调基因缺乏细胞类型特异性(图S9A),而从 SpotGF 去噪数据中识别出的上调基因相比原始数据和 Magic 结果都表现出更高的特异性(图S9B)。
这些结果证明了 SpotGF 去噪流程的有效性:它不仅成功过滤掉了“无效基因”,而且在识别高质量的潜在标记基因方面也表现出色。
同样地,在“子叶-整体维管”细胞类型中的维管细胞中,我们发现 从 SpotGF 去噪数据中识别出的上调程度最高的基因,展现出了最高的细胞类型特异性(图S10A)。这些上调基因的 UMI 计数表达分布也进一步证实了它们在维管细胞中的特异性(图S10B 和图S10C)。此外,原始数据中识别出的上调程度最高的基因中包含了一些扩散效应非常严重的基因,而从 SpotGF 去噪数据中识别出的上调基因则特异地在维管细胞中或紧邻维管细胞的位置表达(图S11A-B)。
类似地,从 SpotGF 去噪数据中识别出的上调基因不仅表达量高,而且对海绵细胞(spongy cells)也具有特异性(图S12A-C),这一点进一步通过空间表达分析得到验证,显示出海绵细胞具有明显的聚集特征(图S13A-B)。总之,这些结果一致表明,从 SpotGF 去噪数据中识别出的上调基因具有更高的准确性。
结果5:SpotGF 有助于提升聚类分析效果与细胞类型推断准确性
我们进一步利用三个公开的 10X Visium 兰花花序数据集 (见表 S5),验证了 SpotGF 对细胞聚类准确性和细胞类型推断的影响。在这些数据中,我们以 H&E 染色图像所衍生的组织位置信息作为真实参考标准,并将原始数据与经过 SpotGF、Magic、SpotClean、Sprod 和 STAGATE 五种方法去噪后的数据,分别聚类为 30 个群集。结果显示,SpotGF 的聚类效果是最优的(图 S14A)。
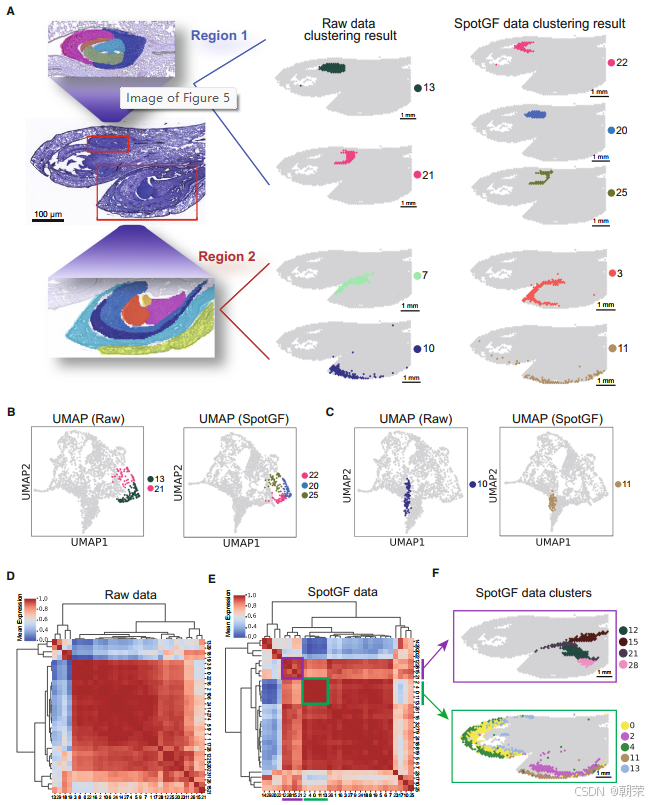
在 花区域一 中,原始数据的聚类结果仅识别出了整个花蕾(群集 13)和分生组织(群集 21)。相比之下,SpotGF 去噪数据的聚类结果成功识别出了更多区域,包括花被片区域(群集 22)、花柱与唇瓣区域(群集 20)以及分生组织(群集 25)。而在 花区域二 中,原始数据的聚类结果仅识别出了外花被片(群集 7)和苞片(群集 10)区域,这些结果与真实解剖结构并不一致。相反,SpotGF 去噪数据的聚类结果成功捕捉到了外花被片组织(群集 3)和苞片组织(群集 11),与真实情况高度吻合(图 5A)。
通过 UMAP(均匀流形近似与投影,用于降维可视化)图 可以看出,SpotGF 去噪数据在每个细胞群中识别出了更多的细胞类型,并且在苞片组织中呈现出更加清晰的细胞聚集现象(图 5B、图 5C 和图 S14A)。我们进一步采用 成对群集-群集相关性分析,对 SRT 数据中不同细胞类型的表达谱进行了比较(图 5D 和图 5E)。结果显示,SpotGF 方法所得到的细胞类型聚类模式更加紧密且彼此独立,尤其对于空间上相邻的细胞类型表现更佳。
具体来说,在层次聚类结果中,群集 12、15、21 和 28 较为相似,它们均对应于兰花组织切片中相邻的尾细胞;而 群集 0、2、4、11 和 13 则紧密关联,代表的是外/内花被片组织中的细胞类型(图 5F)。这些结果说明,SpotGF 通过过滤掉噪声基因,可以准确地识别出细胞亚型。
此外,在使用与原始数据相同的 Scanpy 参数时,SpotGF 去噪数据比原始数据更接近真实解剖结构,尤其是在第二张切片的外花被片组织中表现更为明显(图 S14B)。同样地,SpotGF 在第三张切片的内花被片和蕊喙组织(rostellum tissues)的聚类分析中也表现更优(图 S14C)。总体而言,SpotGF 在多个数据集上始终能产生准确的聚类结果,有效提升了细胞类型识别的准确性。
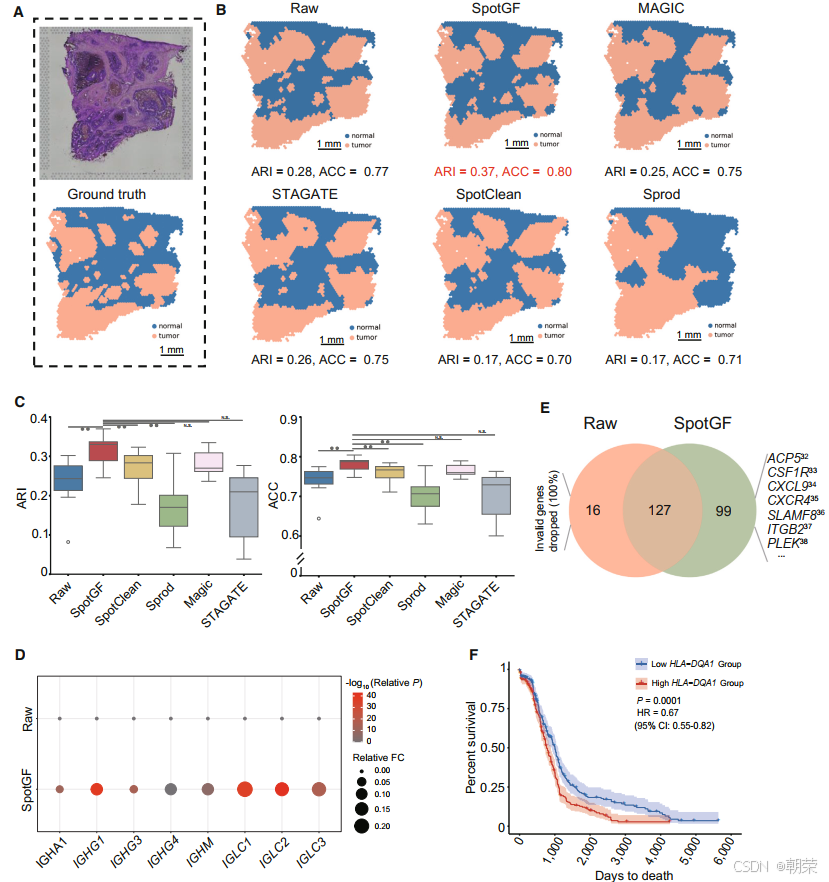
结果6:SpotGF 提高了在人类结直肠癌中识别肿瘤细胞的准确性
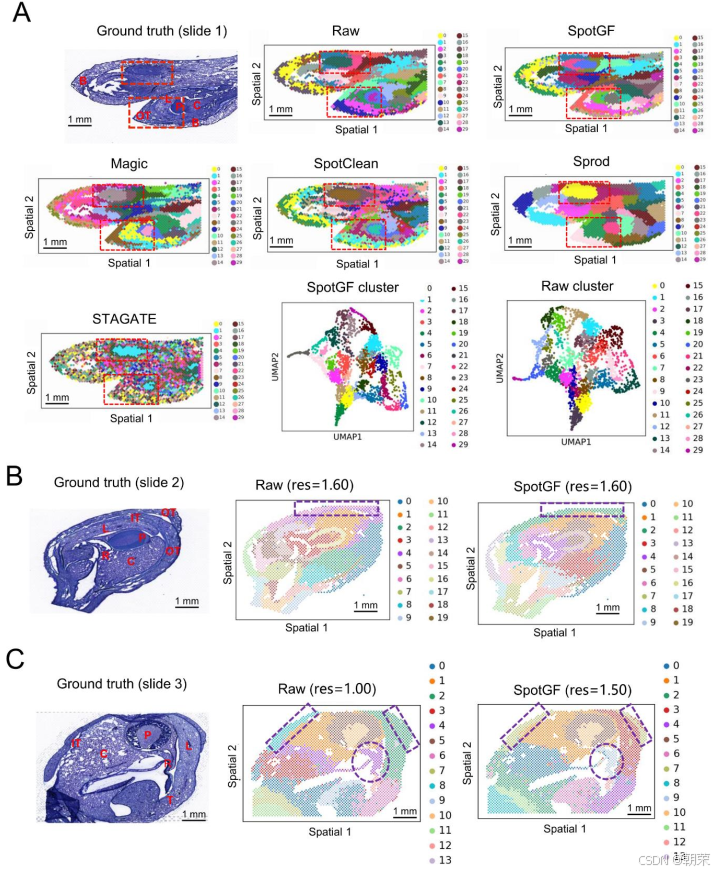
空间分辨转录组学(SRT)技术为将病理图像与基因表达数据相结合提供了独特的机会,有助于提升肿瘤诊断水平并实现精准治疗。我们在 人类结直肠癌数据集(图 6A)上测试了 SpotGF,并将其与其他四种去噪算法进行了对比。我们使用 SpaGCN 对结直肠癌数据集进行了聚类,并对 SpotGF 与其他方法采用了相同的参数设置。
在二分类任务(即从组织中识别肿瘤细胞)中,SpotGF 去噪数据(ARI = 0.37,ACC = 0.80)的准确率是最高的(图 6B 和表 S6)。此外,通过 桑基图(Sankey diagram) 可以看出,SpotGF 去噪数据与真实标签之间在肿瘤细胞上的数据流误差最小(图 S15A)。为了更好地区分肿瘤细胞与正常细胞,我们探索了 12 组精心选择的参数配置,这些配置在保证高预测准确率的同时避免过拟合。在所有评估指标上,SpotGF 的表现均优于原始数据,并且在大多数情况下优于其他四种方法(图 6C、图 S15B 和图 S15C)。
此外,我们基于网格采样方法,在组织中合成了 3,000 个均匀分布的噪声基因(图 S16A),并发现 SpotGF 去噪数据取得了最高的 ARI 值(0.294)和 ACC 值(0.771)(图 S16B)。在相同条件下,我们分别使用 Scanpy、SpaGCN 和 BayesSpace对这五个去噪数据集进行了聚类,结果一致表明 SpotGF 作为去噪预处理算法具有更优的表现(图 S17A–C)。综合来看,这些结果证明 SpotGF 能够提高在人类结直肠癌中识别肿瘤细胞的准确性。
另外,我们还为六个数据集生成了每个细胞中 UMI 计数的热图,结果显示 SpotGF 去噪数据的分布与原始数据非常接近,而其他四种去噪方法则改变了原始数据的表达分布特征(图 S18A)。这些结果进一步支持了 SpotGF 能够特异性地过滤掉具有广泛表达模式的基因。
SpotGF 提升了肿瘤相关上调基因的比例与特异性
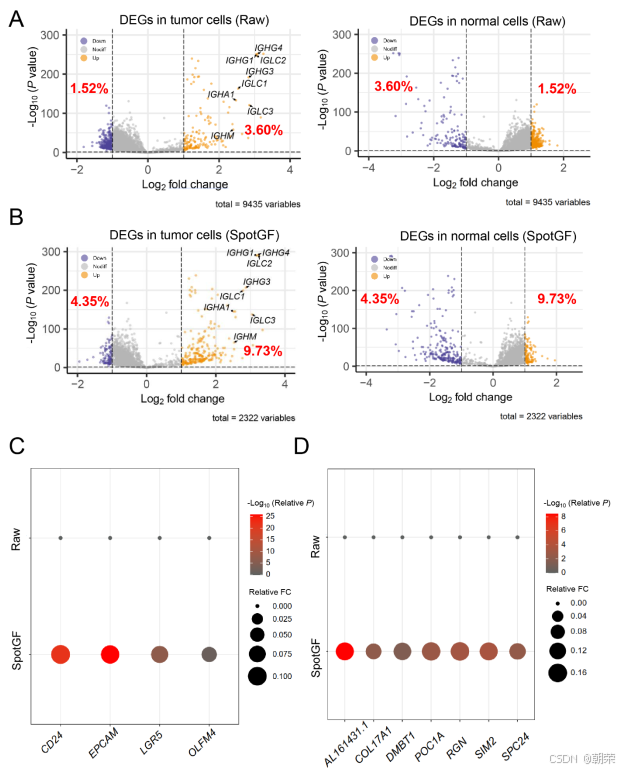
此外,在 SpotGF 去噪数据中,肿瘤细胞内上调的肿瘤相关基因比例(9.73%)高于原始数据(3.60%)(图 S19A、图 S19B 和表 S7)。我们筛选出了 8 个已报道的在肿瘤细胞中上调的基因 [8],发现这些基因在 SpotGF 去噪数据中具有更高的倍数变化(fold change)和更低的 P 值(图 6D)。类似的结果也出现在 7 个正常细胞中的上调基因 以及 CellMarker 数据库 中的 4 个肿瘤细胞标记基因 上(图 S19C 和图 S19D)。
更重要的是,SpotGF 还能够识别出原始数据中未能检测到的额外肿瘤相关基因。例如,在 仅通过 SpotGF 去噪数据识别出的 99 个上调基因中,有 37 个基因的表达与结直肠癌患者较差的生存率呈正相关(根据 TCGA 数据库中的 COAD 数据集 ,P 值 < 0.05)(图 6E)。在这 37 个基因中,有 16 个基因是已知的能够促进结直肠癌细胞增殖、迁移和侵袭过程的基因(表 S8)。此外,剩余的 21 个基因(占 57%)为首次被发现的新基因。在这 37 个基因中,新增的 HLA-DQA1 基因具有最低的 P 值和风险比(HR),提示其可能在肿瘤免疫应答中发挥作用(图 6F)。与之形成对比的是,仅在原始数据中识别出的 16 个上调基因,全部被判定为噪声基因,并被 SpotGF 成功过滤掉(图 6E)。这些发现表明,SpotGF 不仅提高了上调基因的特异性,增强了识别肿瘤细胞的准确性,还促进了肿瘤细胞中潜在标记基因的发现。
讨论
空间分辨转录组学(SRT)数据中的噪声问题与 SpotGF 的解决方案
由于必要的实验操作步骤以及液体实验环境的影响,SRT 数据中捕获的部分基因并不能准确反映其原位(in-situ)表达情况。因此,这些基因失去了其原本的空间特异性,成为不可忽视的噪声(图 1D–F 和图 2A)。这种复杂的空间噪声会对下游分析造成显著影响,导致较大的偏差和错误的结论(图 1F 以及图 S2A–C)。
目前的一些解决方案,如 SpotClean 和 Sprod,主要通过修改每个位点(spot)内的 UMI 计数来实现 SRT 数据的去噪。然而,这些方法在拟合噪声时受到其所选统计模型本身局限性的约束,容易导致额外的假阳性结果,并使那些具有空间特异性的低表达基因被掩盖。
SpotGF:通过过滤“无效基因”实现 SRT 数据去噪
为了克服这一挑战,我们开发了 SpotGF,通过过滤“无效基因”来对 SRT 数据进行去噪。SpotGF 通过构建源分布(source distribution)和目标分布(target distribution),并利用迭代过程计算最优传输方案(optimal scheme)(图 2C)。在评估每个基因的扩散程度时,SpotGF 通过计算该基因在最优方案中的传输成本(transportation cost),进而筛选出那些传统高变异基因(HVGs)计算方法(如方差计算)无法区分的“无效基因”(图 1G)。
此外,我们已经证实,与直接评估空间自相关性(如 Moran's I 值 )相比,SpotGF 能更有效地表征基因的扩散程度。SpotGF 的一大优势在于,即使每个基因的表达位点数量不同,它依然能够准确评估单个基因的扩散系数(图 S3A–C)。与 Magic、SpotClean、Sprod 和 STAGATE 相比,SpotGF 在 兰花数据(图 S20A–L)和结直肠癌数据集(图 S21A–K) 上展现出了更优的运行时间和内存性能。这些广泛的验证结果证明了 SpotGF 在 SRT 数据去噪方面具有更优越的性能。
SpotGF 作为识别空间变异基因(SVGs)的强大工具
此外,SpotGF 已被证明是一种识别空间变异基因(Spatially Variable Genes, SVGs)的强有力工具。我们观察到,SpotGF 评分较高的基因往往具有更高的空间聚集性,表明其有潜力作为 SVGs 被应用。尽管我们的研究在刻画基因扩散程度上取得了进展,但我们也认识到,SpotGF 评分的自动阈值选择方法仍存在一定局限性。将某个基因判定为“无效基因”需要仔细综合考量组织特异性信息、扩散程度以及 SRT 数据中固有的生物学信息。我们目前的自动化方案倾向于过滤掉大量无效基因,因此我们建议在后续的聚类分析中使用所有保留下来的基因。同时,我们还设计了额外的接口,支持用户根据自身数据特点自定义阈值。我们将在未来的研究中继续深入探索这一复杂问题。
SpotGF:提升 SRT 数据质量,助力精准生物学解读
尽管空间分辨转录组学(SRT)数据在解决生物学问题方面已获得广泛应用并展现出巨大潜力,但该领域仍需更加关注 SRT 数据的质量问题。SpotGF 去噪算法是专门为 SRT 数据量身定制的,是 SRT 数据处理流程中至关重要的第一步。通过有效降低 SRT 数据中的噪声,SpotGF 为下游分析奠定了坚实基础,有助于更准确地解读潜在的生物学现象。