【工具】metaTP:一种集成了自动化工作流程的元转录组数据分析工具包

介绍
Background
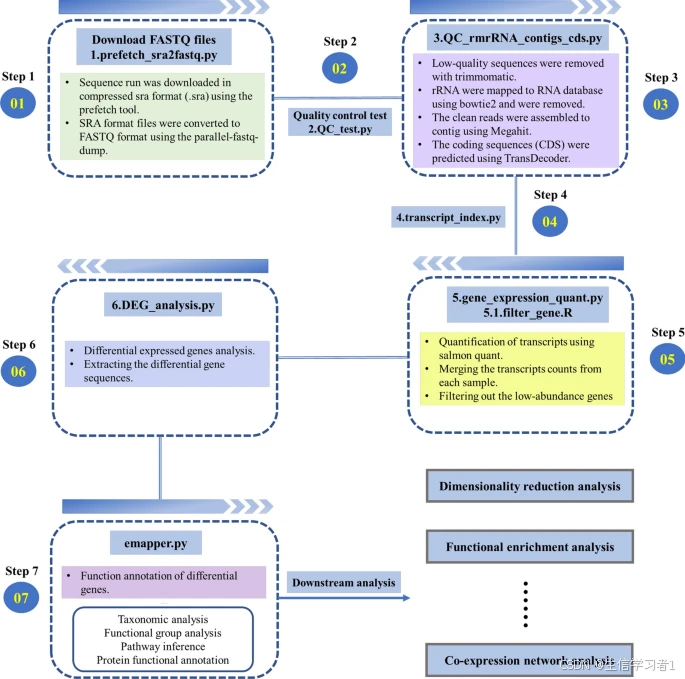
The accessibility of sequencing technologies has enabled meta-transcriptomic studies to provide a deeper understanding of microbial ecology at the transcriptional level. Analyzing omics data involves multiple steps that require the use of various bioinformatics tools. With the increasing availability of public microbiome datasets, conducting meta-analyses can reveal new insights into microbiome activity. However, the reproducibility of data is often compromised due to variations in processing methods for sample omics data. Therefore, it is essential to develop efficient analytical workflows that ensure repeatability, reproducibility, and the traceability of results in microbiome research.Results
We developed metaTP, a pipeline that integrates bioinformatics tools for analyzing meta-transcriptomic data comprehensively. The pipeline includes quality control, non-coding RNA removal, transcript expression quantification, differential gene expression analysis, functional annotation, and co-expression network analysis. To quantify mRNA expression, we rely on reference indexes built using protein-coding sequences, which help overcome the limitations of database analysis. Additionally, metaTP provides a function for calculating the topological properties of gene co-expression networks, offering an intuitive explanation for correlated gene sets in high-dimensional datasets. The use of metaTP is anticipated to support researchers in addressing microbiota-related biological inquiries and improving the accessibility and interpretation of microbiota RNA-Seq data.Conclusions
We have created a conda package to integrate the tools into our pipeline, making it a flexible and versatile tool for handling meta-transcriptomic sequencing data. The metaTP pipeline is freely available at: https://github.com/nanbei45/metaTP.
背景信息
测序技术的普及使得元转录组学研究能够在转录水平上更深入地理解微生物生态。对组学数据的分析涉及多个步骤,需要使用各种生物信息学工具。随着公共微生物组数据的日益丰富,进行元分析可以揭示有关微生物组活动的新见解。然而,由于样本组学数据处理方法的差异,数据的可重复性往往受到损害。因此,开发高效的分析工作流程至关重要,以确保微生物组研究中的可重复性、可再现性和结果的可追溯性。
结果
我们开发了 metaTP 这一工具,它是一个集成了多种生物信息学工具的流程,用于全面分析元转录组数据。该流程包括质量控制、非编码 RNA 去除、转录表达量量化、差异基因表达分析、功能注释以及共表达网络分析。为了量化 mRNA 表达量,我们依靠使用蛋白质编码序列构建的参考索引,这有助于克服数据库分析的局限性。此外,metaTP 还提供了一个计算基因共表达网络拓扑特性的功能,为高维数据集中相关基因集提供了直观的解释。使用 metaTP 预计将有助于研究人员解决与微生物群相关的生物学问题,并提高微生物群 RNA-Seq 数据的可访问性和解读性。
结论
我们已创建了一个 conda 包,将这些工具整合到我们的流程中,使其成为处理元转录组测序数据的灵活且多功能的工具。metaTP 流程可在以下网址免费获取:https://github.com/nanbei45/metaTP 。
代码
https://github.com/nanbei45/metaTP
参考
- metaTP: a meta-transcriptome data analysis pipeline with integrated automated workflows
- https://github.com/nanbei45/metaTP