16S18S_分析步骤(2)
1.概述
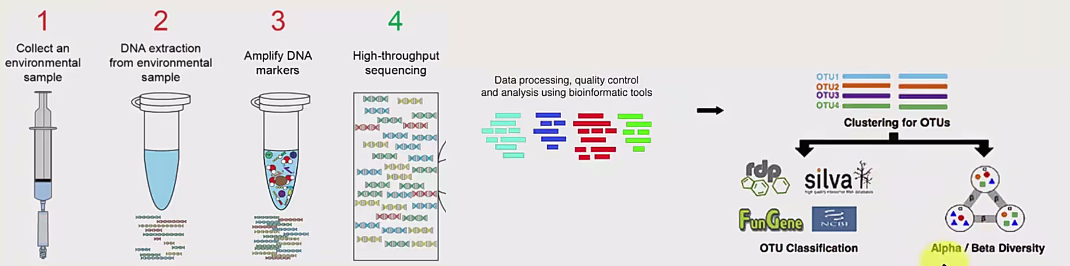
步骤1:收集环境样本
- 操作:从环境中收集样本,这些样本可能包括土壤、水体、空气、人体微生物群等。
- 目的:获取包含目标微生物的样本,以便进行后续的DNA提取和分析。
步骤2:从环境样本中提取DNA
- 操作:使用化学和/或物理方法从收集的样本中提取总DNA。
- 目的:获取纯净的DNA,以便进行PCR扩增和测序。
步骤3:扩增DNA标记
- 操作:使用特定的引物对16S rRNA基因的V3和V4区域进行PCR扩增。
- 目的:增加目标DNA片段的数量,使其达到高通量测序所需的浓度。
步骤4:高通量测序
- 操作:利用高通量测序技术(如Illumina平台)对扩增的DNA片段进行测序。
- 目的:生成大量的DNA序列数据,用于后续的生物信息学分析。
数据处理和分析
- 操作:使用生物信息学工具对测序数据进行处理、质量控制和分析。
- 目的:从原始测序数据中提取有用的信息,如OTU(操作分类单元)的分类和多样性。
后续分析
- OTU聚类:将相似的序列聚类为OTUs,每个OTU代表一个微生物物种或分类单元。
- OTU分类:使用数据库(如RDP、SILVA、FunGene)对OTUs进行分类,确定其分类地位。
- 多样性分析:计算α多样性(样本内多样性)和β多样性(样本间多样性),评估微生物群落的多样性和结构。
文库构建
-
DNA提取:
- 首先从环境样本中提取总DNA,这可能包括土壤、水体、空气或人体样本等。
-
设计和使用通用引物:
- 设计包含V3和V4区域特异性序列的通用引物,这些引物能够识别并结合大多数细菌的16S rRNA基因的这些区域。
-
PCR扩增:
- 使用设计的通用引物对提取的DNA进行PCR扩增,以增加V3和V4区域的拷贝数,为后续的测序准备足够的模板。
-
纯化扩增产物:
- 扩增结束后,需要对PCR产物进行纯化,以去除未结合的引物、dNTPs和其他PCR副产物,确保后续步骤的准确性。
-
加入接头和Index信息:
- 在纯化后的扩增子两端加入测序接头(adapters),这些接头包含了测序平台所需的序列信息,如Illumina平台的P5和P7接头。
- 同时,加入Index信息,这些是独特的序列标签(如Index1和Index2),用于区分不同样本或不同实验条件下的DNA片段。
-
第二轮PCR:
- 通过第二轮PCR将接头和Index信息固定到扩增子片段上。这一步确保了每个扩增子都携带了必要的测序信息和样本标识。
-
文库构建完成:
- 第二轮PCR结束后,得到的DNA片段即为构建好的测序文库,可以直接用于高通量测序。
-
文库质量检查:
- 在测序前,通常需要对构建好的文库进行质量检查,如使用琼脂糖凝胶电泳评估文库的浓度和大小分布,或使用生物分析仪进行更精确的质量评估。
-
测序:
- 将质量合格的文库上机测序,获取大量的16S rRNA基因序列数据。
fastq数据

第一行:序列标识符(ID)
- 格式:以 “@” 开头,后跟一串字符,表示该条序列的唯一标识符。
- 内容:这串字符通常包含了关于序列的来源、样本信息、测序平台等元数据。
- 特点:在同一份FASTQ文件中,每个序列的标识符都是唯一的,即使在不同的文件中也不会重复。
第二行:碱基序列
- 格式:由A、C、G、T和N五个字符组成,表示DNA序列。
- 内容:这一行包含了实际的DNA序列信息,其中:
- A、C、G、T:分别代表腺嘌呤(Adenine)、胞嘧啶(Cytosine)、鸟嘌呤(Guanine)和胸腺嘧啶(Thymine)。
- N:代表无法识别的碱基,可能是由于测序过程中的不确定性或错误。
- 特点:这一行是FASTQ文件的核心,包含了实际的生物学信息。
第三行:分隔符
- 格式:以 “+” 开头,后跟一个空格或直接结束。
- 内容:这一行在现代FASTQ文件中通常为空,用于分隔序列信息和质量分数。
- 历史背景:在早期的FASTQ格式中,这一行可能包含与第一行相同的序列标识符,用于验证序列信息的完整性。但在现代应用中,这一行通常被省略,以节省存储空间。
第四行:质量分数
- 格式:由一系列数字组成,每个数字对应第二行中的一个碱基。
- 内容:这一行描述了每个碱基的测序质量,通常使用Phred质量分数表示。
- Phred质量分数:是一种对碱基测序质量的量化表示,分数越高,表示碱基识别的准确性越高。Phred分数通常转换为ASCII字符表示,以便于存储和处理。
- 特点:质量分数对于后续的数据分析至关重要,如序列比对、变异检测等,因为它们提供了关于数据可靠性的重要信息。
原始测序数据处理
原始测序数据处理
1. 根据测序barcode(index)序列区分不同的样本序列
- 操作:在测序过程中,每个样本的DNA片段都会附加上一个独特的barcode序列,这个序列作为样本的标签。
- 目的:通过识别这些barcode序列,可以将原始测序数据中混合的序列拆分到对应的样本中,实现样本的区分。
2. 去除接头序列,过滤低质量序列
- 操作:原始测序数据通常包含接头序列和低质量的序列。
- 去除接头序列:接头序列是PCR过程中添加的,用于测序的识别和结合,需要在数据分析前去除。
- 过滤低质量序列:低质量序列可能包含较多的测序错误,影响后续分析的准确性,需要被过滤掉。
- 目的:确保后续分析的数据质量,提高分析结果的可靠性。
3. 双端测序序列的拼接
- 操作:对于双端测序(paired-end sequencing)的数据,需要将两端的序列通过序列之间的overlap(重叠区域)拼接成单条序列(Tags)。
- 工具:此步骤通常由专门的软件如flash完成。
- 目的:拼接后的序列更长,包含的信息更丰富,有助于提高物种鉴定的准确性。
序列1(来自片段的一端):
ATCGTACGTAGCTAGCTAGCTACGTAGCTACG
序列2(来自片段的另一端):
TAGCTAGCTAGCTAGCTAGCTACGTAGCTAG
步骤1:识别重叠区域
我们首先需要找到这两个序列之间的重叠部分。在这个例子中,重叠区域是“TAGCTAGCTAGCTA”,长度为12bp(碱基 对)。
步骤2:序列拼接
接下来,我们将两个序列通过重叠区域拼接起来。由于两个序列是直接连续的,我们可以直接将序列1与序列2的剩余部分连接起来:
拼接后的序列:
ATCGTACGTAGCTAGCTAGCTACGTAGCTAGTAGCTAGCTAGCTACGTAGCTAG
这里,我们从序列1的开始到重叠区域结束,然后紧接着序列2的重叠区域之后的剩余部分。
4. 去除嵌合体序列
- 操作:嵌合体序列是指由两个或多个不同来源的DNA片段错误拼接而成的序列。
- 目的:嵌合体序列不是真实的生物序列,去除这些序列可以避免对微生物多样性分析结果产生误导。
嵌合体
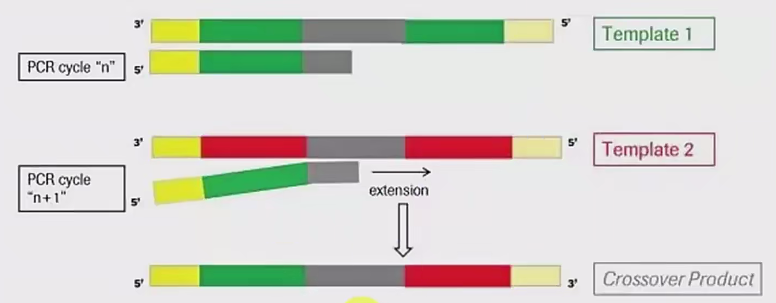
嵌合体的形成
-
形成过程:
- 在图示中,DNA聚合酶开始时在Template 1上合成新的DNA链(绿色部分),然后可能由于模板DNA的退火不完全或引物的非特异性结合,聚合酶从Template 1上脱离并切换到Template 2上继续延伸(红色部分)。
- 这种在不同模板之间切换的延伸过程导致了一个嵌合体DNA分子的产生,这个分子包含了来自两个不同模板的DNA序列片段。
-
结果:
- 嵌合体DNA分子(Crossover Product)在图中用不同颜色表示,显示了它是由两个不同模板的DNA片段拼接而成的。