剑桥大学最新研究:基于大语言模型(LLM)的分子动力学模拟框架,是MD的GPT时刻还是概念包装?
近期,剑桥大学 Michele Vendruscolo 团队在预印本平台上发布了一项最新研究,提出了一个名为 MD-LLM 的框架,旨在为高效研究蛋白质动态提供一种全新的思路。简单来说,他们希望借助大语言模型(LLM),在只有单一构象状态数据的情况下,就能预测蛋白质的其他构象状态。
结果速览
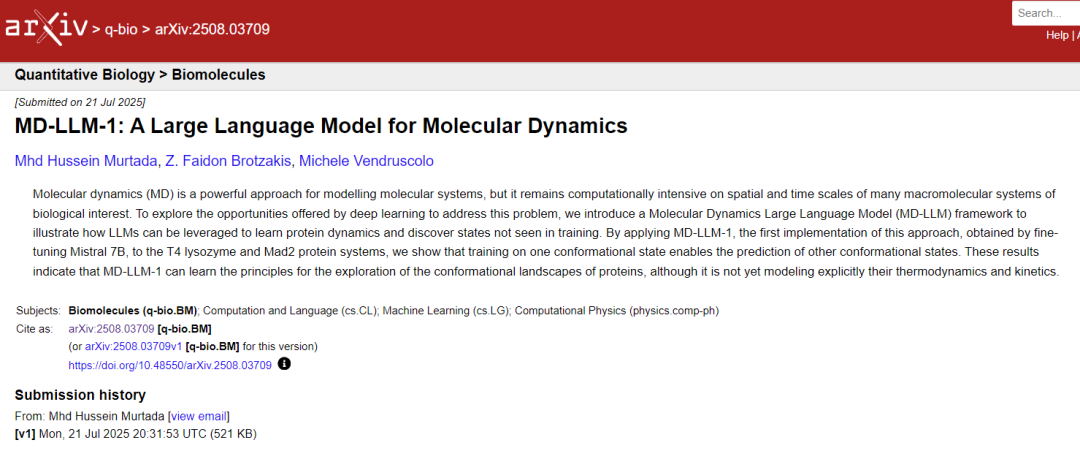
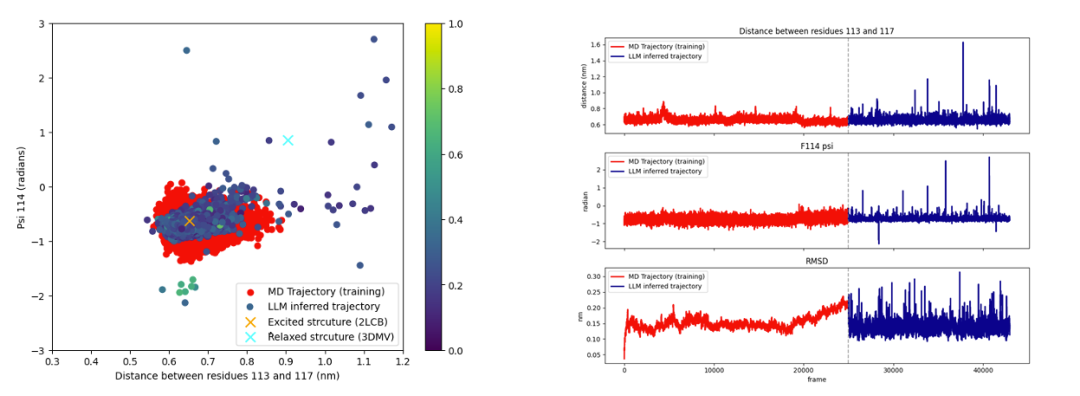
在技术路径上,该研究团队①首先通过 FoldToken 将蛋白质三维构象编码为离散 token,使其能够被大语言模型处理;②随后,在 Mistral 7B 模型(一种大语言模型)的基础上进行微调,让模型学会从单一构象生成其他可能的状态。③在推理阶段,他们采用滚动窗口策略,并结合温度(temperature)、top-k 等采样参数,生成在几何上看似合理的构象。实验显示,在 T4 溶菌酶和 Mad2 蛋白上,这种方法能在只见过 native 态的情况下,生成 excited 态结构。尤其在 T4 溶菌酶 L99A 突变体的实验中,模型甚至能“发现”低丰度状态,体现出一定的跨状态预测能力。
优势
总体来说,MD-LLM 为分子动力学开辟了一条新路。它将 LLM 创新性地引入这一领域,突破了传统基于牛顿第二定律的分子动力学模拟在时间尺度上的限制。更令人惊讶的是,它并未显式嵌入物理化学知识,却依然能在短时间内生成多种可能构象,这无疑为蛋白质结构预测和稀有状态发现带来了新想象空间。
存在的隐患
但优势的背后,也潜藏着明显的风险。首先,这个框架缺乏普适性,即更换蛋白体系就必须重新微调模型,使用门槛不低。其次,由于不基于物理规律,生成的构象很难判断究竟是真实存在,还是 LLM“幻觉”的产物。更麻烦的是,模型生成的不同构象之间缺乏明确的动力学关系,无法像传统模拟那样给出连续、可追溯的状态演化路径。换句话说,即便结果在三维空间里看上去合理,我们也无法确认它在真实物理世界中站得住脚。
更进一步地思考
问题的根源,其实在于大语言模型本身的局限。LLM 的长处在于模式匹配和生成速度,而不是对底层物理过程的理解。数学家陶哲轩提到,AI 的发展高度依赖经验、算力和数据,这让成功难以复制、失败难以解释。他将这种状态比作“炼金术”,并强调理论才是技术长期进步的核心驱动力。当下的 AI 缺乏类似压缩感知那样成体系的理论基础,GPT 等大模型的惊艳表现更多是资源堆砌和反复试错的结果,而非建立在坚实数学原理之上。黑箱模型的不可解释性不仅限制了科学研究中的可验证性,也在安全与可控性方面构成挑战。要让 AI 真正可靠,就必须回到理论的根基,构建清晰、可验证的数学模型,从而摆脱“大力出奇迹”的发展模式。

放在这个背景下,MD-LLM 的不足就不难理解了。它依赖的依然是模式生成,而不是物理推演,缺少理论保证和物理约束,这让它在低容错率的科学任务中难以独当一面。在科研实践中,LLM 或许可以在一分钟内给出结果,但研究者往往要花上数倍甚至数十倍的时间去验证其正确性,这种高验证成本几乎抵消了生成速度的优势。也正因如此,尽管 MD-LLM 展示了 LLM 在结构生物学中的巨大潜力,我依然倾向于认为,经典的、基于牛顿第二定律的分子动力学模拟不会在 LLM 时代被淘汰。至少在当下,LLM 更适合作为假设生成和探索性分析的工具,而不是取代物理定律驱动的核心模拟方法。
个人观点,仅供参考。
欢迎大家在评论区留下你的看法,理性讨论、友善发言