【数据分析】宏基因组荟萃分析(Meta-analysis)的应用与实操指南
禁止商业或二改转载,仅供自学使用,侵权必究,如需截取部分内容请后台联系作者!
文章目录
-
- 介绍
-
- 一、宏基因组(microbiome)荟萃分析的现状
- 二、待解决的核心难题
- 三、解决痛点
- 四、原理与方法流程
- 加载R包
- 导入数据
- 数据预处理
- 荟萃分析
- 荟萃分析系数
- 总结
- 系统信息
介绍
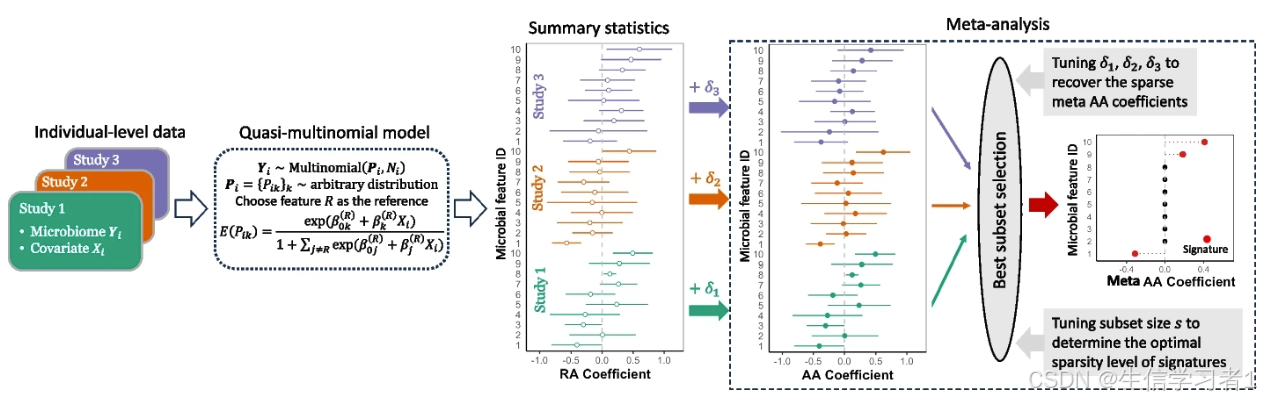
宏基因组荟萃分析是把多项独立宏基因组研究联合起来,系统评估微生物特征(菌种、基因、通路等)与宿主表型或暴露因素之间关联的统计学方法。它通过扩大样本量、整合跨人群异质性,提高发现稳健微生物标志物的能力。传统方法直接套用 GWAS 或临床 meta-analysis 时,会忽视微生物数据的“成分性、深度不均、零膨胀、批次效应”等独特属性,导致假阳性、不可重复。新兴工具如 Melody 在保留原始计数、无需归一化或零填充的前提下,为每项研究生成成分性-感知的汇总统计量,再跨研究统一模型框架,识别在所有队列中均与宿主变量存在绝对丰度关联的“驱动菌”,从而实现可泛化、高可信度的微生物组发现。
一、宏基因组(microbiome)荟萃分析的现状
- 数据爆炸:高通量测序让“无培养”研究微生物组成为可能,但单个研究样本量小,结果难以泛化。
- 汇总需求:近几年大量研究开始尝试 meta-analysis 以扩大样本量、提高统计功效 [2–5]。
- 技术瓶颈:传统 meta-analysis 策略(如 GWAS 或临床常用的固定/随机效应模型)直接套用到微生物组数据时,表现不佳,难以发现稳定、可重复的“微生物标志物”。