DeePNAP:一秒预测蛋白-DNA/蛋白-RNA结合强度
字数 705,阅读大约需 4 分钟
DeePNAP 是一种基于机器学习的模型,能仅从蛋白质和核酸(DNA或RNA)序列,精确预测蛋白-核酸相互作用(PNAIs)的结合亲和力(如解离常数 KD )和突变导致的自由能变化ΔΔG。它使用 ProNAB 数据库中 14401 个条目训练,在预测 KD 时,平均皮尔逊相关系数约 0.86,均方根误差 0.83;集成模型预测 KD 的相关系数达 0.93,均方根误差 0.63 ,在同类工具中表现出色。
在线使用地址:http://14.139.174.41:8080/input_page
快速上手
按照下图输入蛋白序列信息、核酸序列信息。如果你需要探究蛋白上的位点突变对结合核酸的能力影响,还可以输入突变信息,这样你可以得到突变后蛋白质与核酸的结合自由能变化(ΔΔG)
注意:
①核酸序列可以是DNA也可以是RNA;
②该程序暂时还不支持区分dsDNA和ssDNA,即无论是哪种情况,只需输入一条5’→3’方向的DNA序列。
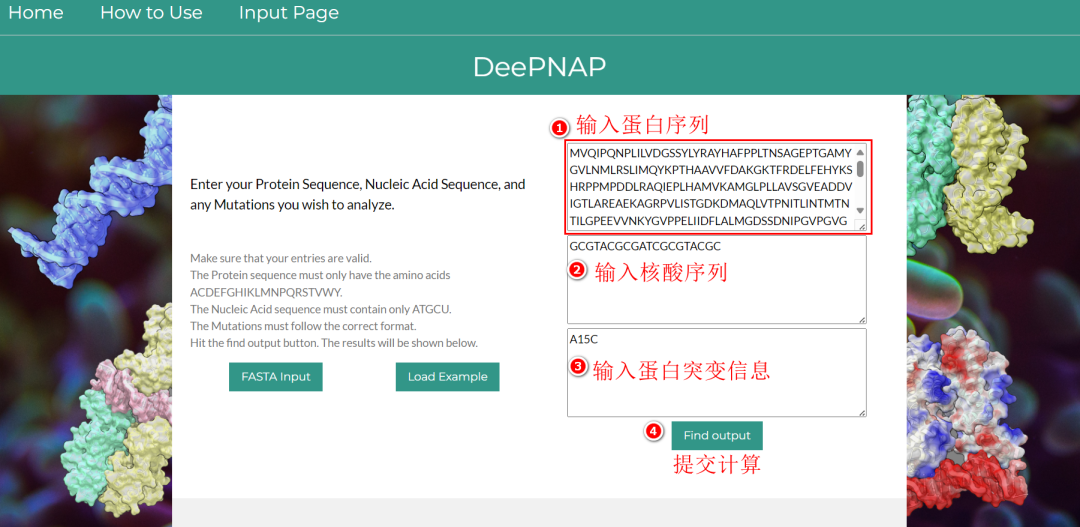
另外,蛋白突变信息的输入需要遵循一定的语法,详细介绍如下:
-
1. 单氨基酸删除
-
格式:del + 氨基酸 + 位置。
-
示例:删除序列中第 3 位的 C →
delC3
-
2. 单氨基酸替换为多氨基酸序列
-
格式:原氨基酸 + 位置 + 新氨基酸序列。
-
示例:将第 5 位 V 替换为 TUV →
V5TUV
-
3. 多氨基酸删除(序列删除)
-
格式:del + 起始氨基酸 + 起始位置 + "-" + 终止氨基酸 + 终止位置。
-
示例:删除第 2 位 T 到第 9 位 V 的序列 →
delT2-V9。
-
4. 单氨基酸替换
-
格式:原氨基酸 + 位置 + 新氨基酸。
示例:将第 3 位 C 替换为 T →C3T。
-
5. 多个突变
-
多个突变:用英文逗号 , 分隔不同突变指令。
delT17,V5TUV
结果展示
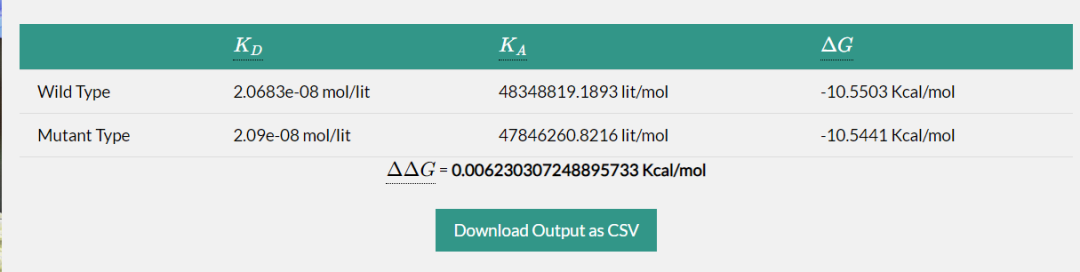
上一步提交计算后,可以秒得结果。往下滑动可以看到如下内容:
上图结果之间的表达式关系如下所示:
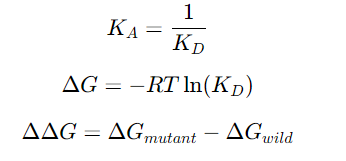
-
KD:解离常数。衡量分子间结合强弱的指标,数值越小表示结合越紧密。 -
KA:结合常数。数值越大表示结合亲和力越强。 -
ΔG: 自由能变化。 表示从游离状态到结合状态的能量差异,负值表示结合自发进行。 -
ΔΔG:突变自由能变化。突变前后的自由能差值,用于评估突变对结合强度的影响。例如,当ΔΔG大于0时表示突变导致结合减弱,反之则增强。
参考文献:
Pandey, Uddeshya, et al. "Deepnap: A deep learning method to predict protein–nucleic acid binding affinity from their sequences." Journal of Chemical Information and Modeling 64.6 (2024): 1806-1815.