一台笔记本实现基因表达敲除?!scTenifoldKnk 单细胞基因模拟敲除教程
生信碱移
单细胞基因模拟敲除
scTenifoldKnk 是一种基于单细胞RNA测序数据进行虚拟基因敲除分析的方法,能够用于预测特定基因在某一细胞群体中敲低后的整体基因表达谱变化,也可以扩展到多细胞群体的敲除分析。
基因扰动实验是研究特定基因功能作用的强大方法。常用的方案一般是使用基因改造动物进行的基因敲除(KO)或CRISPR基因扰动。在KO实验中,研究人员一般通过对比KO和野生型WT实验动物的表型或者多组学差异来推断目标基因的功能。
传统的基因敲除实验通常需要大量的实验和动物资源。最近发展的技术,如Perturb-seq,结合了CRISPR扰动和单细胞RNA测序(scRNA-seq)来进行遗传筛选,使得研究人员能够在大量细胞中研究基因功能。
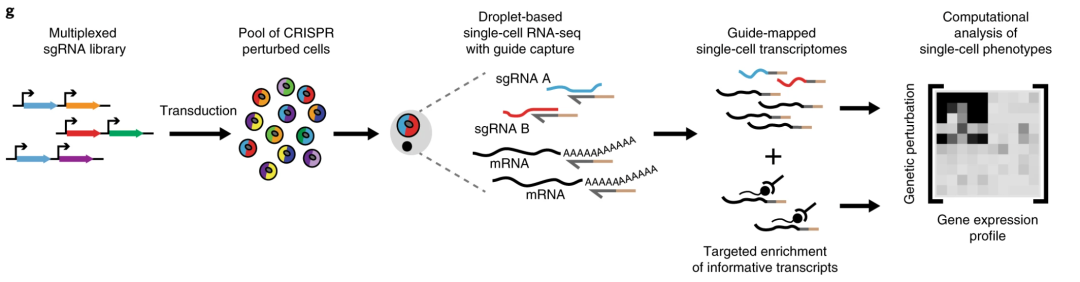
▲ CRISPR-based Perturb-seq原理。该方法可以在成千上万个细胞中同时引入不同的基因扰动,结合单细胞测序系统性地推断基因功能与调控网络。首先,研究者构建包含多种sgRNA(导向RNA)的多重化文库,每种sgRNA针对一个目标基因,通过设计合成并载入特定表达载体,使其在后续实验中可同时对多个基因实施扰动。图中左侧的不同颜色箭头表示不同的sgRNA,每条sgRNA都由特定启动子驱动表达。随后,这一文库被转导入目标细胞群体中,使得每个细胞携带一种或少数几种sgRNA,形成包含多种扰动的细胞混合群。图中彩色的细胞即表示携带不同扰动的异质细胞。接着,利用单细胞RNA测序平台如10x Genomics将单个细胞封装至微液滴中,捕获其转录的mRNA以及所表达的sgRNA标记序列,并进行文库构建与测序。根据前面的描述我们不难知道,由于细胞中的sgRNA与mRNA一起被检测,通过识别每个细胞的sgRNA就可以得知每个细胞的扰动身份(到底是哪些sgRNA转染进入该细胞)。在数据处理中,将每个细胞的基因表达与其对应的sgRNA进行映射,构建出扰动-表达对应关系,从而生成带有基因扰动注释的单细胞转录组数据,进一步的可以研究特定基因功能的变化。DOI: 10.1038/s41587-020-0470-y。
尽管如此,创建大规模的CRISPR文库仍然是一个主要的技术挑战。先前其实已经有了多种工具用于在单细胞水平模拟基因敲除扰动,包括有监督型的工具比如GEARS、scGen、CPA,以及无监督的工具比如scTenifoldKnk、CellOracle、GenKI。前者基于已有的扰动数据集进行监督训练,而后者则基于一些无监督神经网络或者网络建模进行无监督推断。
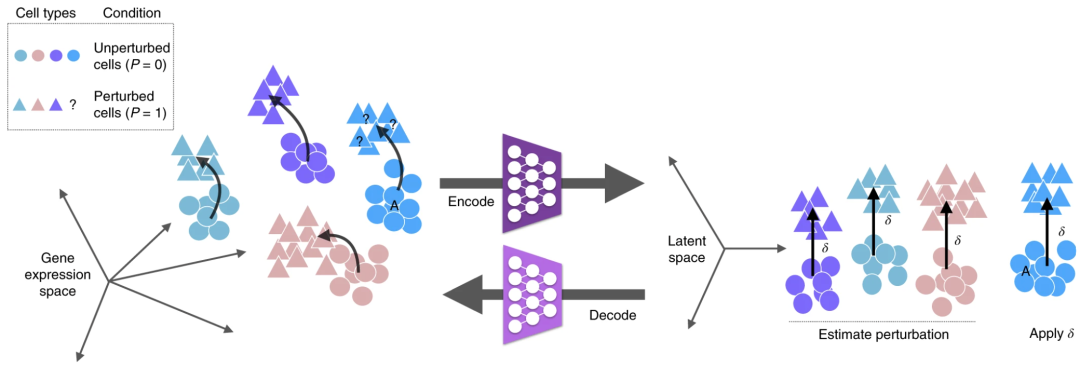
▲ scGen原理(有监督模拟敲除方法)。给定一组在对照组和刺激条件下观察到的细胞类型,scGen的目标是通过训练一个能够学习并泛化训练集中细胞响应模式的模型,来预测新细胞类型 A(蓝色)的扰动反应。在scGen中,该模型采用变分自编码器架构,其预测结果通过自编码器潜在空间中的向量运算获得。具体而言,scGen利用编码器网络将基因表达数据投影至潜在空间,并获取一个代表训练集中扰动与未扰动细胞间潜在空间差异的向量δ。基于δ,scGen对类型A的未扰动细胞在潜在空间进行线性外推。随后,解码器网络将这些潜在空间的线性预测进行重构推演出更新的基因表达。DOI:10.1038/s41592-019-0494-8。
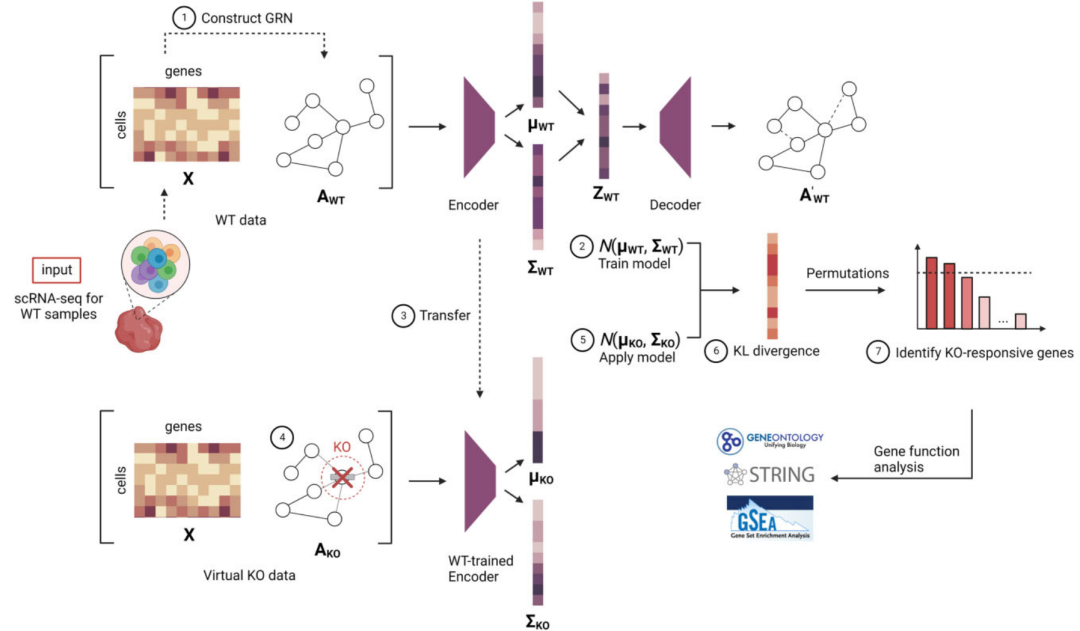
▲GenKI原理( 无监督模拟敲除方法)。GenKI利用任意假定的野生型单细胞RNA测序数据,结合图变分自编码器对基因调控网络进行建模,以实现模拟基因敲除效应的预测分析。首先,GenKI从WT数据中构建调控网络,并使用VGAE学习其潜在空间表示,获得WT条件下的潜在分布参数。随后,保留训练好的编码器,将目标基因从网络中“剪除”以构建虚拟KO网络,并利用相同表达矩阵提取KO状态下的潜在空间表示。通过计算WT与KO潜在表示之间的KL散度,量化基因敲除所带来的系统性扰动,进而通过统计检验识别受影响的KO响应基因,并进行功能注释和通路富集分析。DOI:10.1038/s41592-019-0494-8。
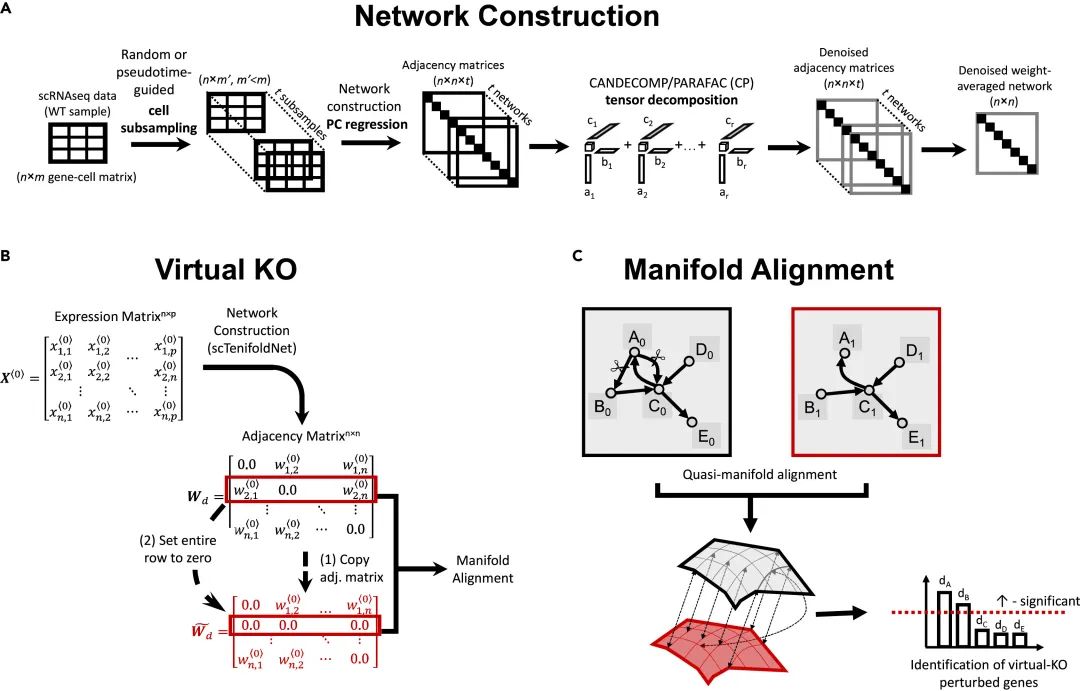
▲ scTenifoldKnk原理( 无监督模拟敲除方法)。scTenifoldKnk 也是一种基于单细胞RNA测序数据进行模拟基因敲除分析的方法,其流程包括三大模块:网络构建、虚拟敲除与流形对齐。首先,从野生型样本的表达矩阵中通过细胞子采样(每次采样生成的矩阵都会进行下方分析)、主成分回归和张量分解构建去噪后的基因调控网络(scGRN,多次采样生成多个子网络);随后,将该网络复制并将目标KO基因对应的调控边设置为零,生成模拟敲除网络;最后,利用流形对齐方法将原始网络与模拟KO网络嵌入到潜在空间中,通过计算基因在两个网络中的表示差异,识别出对敲除敏感的差异调控基因,并据此推断目标基因的潜在功能。DOI:10.1016/j.patter.2022.100434。
考虑到大部分铁子使用的是R语言编辑环境,所以今天小编给大家介绍这些工具中使用R语言的scTenifoldKnk工具,其于2022年发表于Cell子刊 Patterns [IF: 6.7],可以算得上是最早的单细胞无监督基因敲除方法了。这个方法目前有37条被引,其中不含一些大子刊,所以也是值得学习一手的。

scTenifoldKnk的github如下,感兴趣的铁子可以进一步了解:
-
https://github.com/cailab-tamu/scTenifoldKnk
01.安装R包
使用以下代码安装R包:
devtools::install_github("cailab-tamu/scTenifoldKnk")
02.模拟基因敲除
① 相关R包导入:
library(dplyr)
library(Seurat)
library(scTenifoldKnk)
library(ggplot2)
library(ggrepel) # 用于防止标签重叠
② 准备输入数据,这里小编使用一个标准的seurat对象af来提取count矩阵作为输入:
# 查看af对象
af
#An object of class Seurat
#39128 features across 1091 samples within 2 assays
#Active assay: SCT (18305 features, 3000 variable features)
# 1 other assay present: RNA
# 2 dimensional reductions calculated: pca, umap# 提取count矩阵作为输入
countMatrix <- GetAssayData(af, slot = "counts")
③ 执行敲除分析(推荐用高变基因做,不然内存时间双重爆炸):
result <- scTenifoldKnk(countMatrix = countMatrix, gKO = 'GAB1', #需要敲除的基因qc = TRUE,#是否进行QCqc_mtThreshold = 0.1,#mt阈值qc_minLSize = 1000,#文库阈值(细胞测到的基因总数)nc_nNet = 10, #子网络数量nc_nCells = 500, #每个网络中随机抽取的细胞数nc_nComp = 3 #PCA 的主成分数量)
scTenifoldKnk的参数可以查看小编代码中的注释。简单来讲,参数gKO用于指定敲除的基因;qc开头的系列参数能够选择是否对矩阵进行质控以及相应指标;nc开头的参数对应网络的构建,分别包括子网络的数量、使用的PC数量、随机抽取的细胞数量,数值越大运行时间越长但是结果可能会更稳定(具体意义常见上面方法描述)。
④ 结果的可视化,首先来看看排名前20的基因:
top_genes <- head(result$diffRegulation[order(-result$diffRegulation$FC), ], 20)
ggplot(top_genes, aes(x=reorder(gene, FC), y=FC)) +geom_bar(stat='identity', fill='steelblue') +coord_flip() +labs(title="Top 20 Differentially Regulated Genes",x="Gene", y="FC") +theme_minimal()
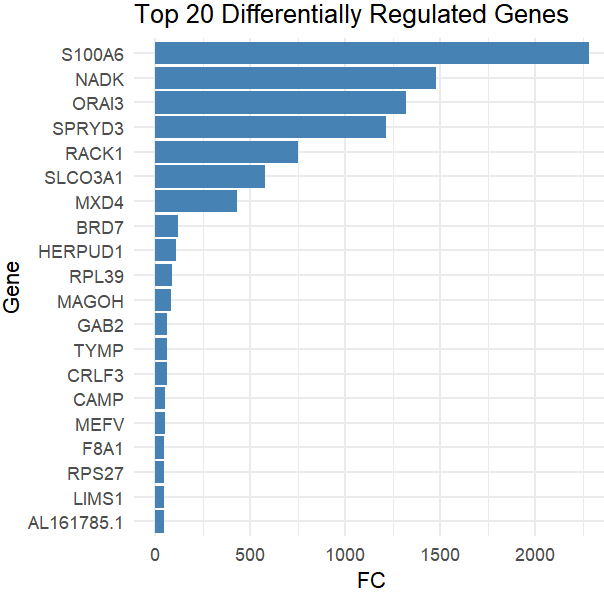
需要注意的是,result$diffRegulation是差异调控分析的结果表,其中展示了在虚拟基因敲除前后,每个基因的发生的潜在变化。每一行对应一个基因,包括以下信息:
-
distance:该基因在两种条件下(WT与模拟KO)位置的欧氏距离,能够反映其调控变化的幅度; -
Z:经过Box-Cox变换后计算的标准化Z分数; -
FC:相对于预期的变化幅度,代表基因变化的倍数; -
两个统计显著值:分别是由卡方分布计算的显著性p值(
p.value)和经过FDR校正后的调整p值(p.adj)。
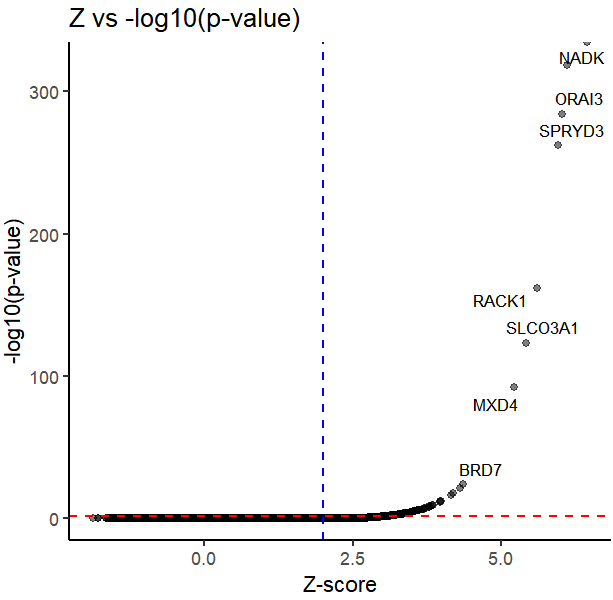
当然,也可以同时绘制Z-score与P值的结果:
df <- result$diffRegulation
df$log_pval <- -log10(df$p.adj)
label_genes <- subset(df, abs(Z) > 2 & p.adj < 0.01)
ggplot(df, aes(x=Z, y=log_pval)) +geom_point(alpha=0.5) +geom_hline(yintercept=-log10(0.05), linetype="dashed", color="red") +geom_vline(xintercept=c(2), linetype="dashed", color="blue") +geom_text_repel(data=label_genes, aes(label=gene),size=3, max.overlaps=50) +labs(title="Z vs -log10(p-value)",x="Z-score", y="-log10(p-value)") +theme_classic()
运行时间还是比较长的
可以做一些初步的验证分析